 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Annals of African Medicine, Vol. 3, No. 2, 2004, pp. 55-62 ROLE OF FREE RADICALS IN PATHOGENESIS OF DIABETES NEPHROPATHY D. S. Mshelia Chemical Pathology Department, University Maiduguri Teaching
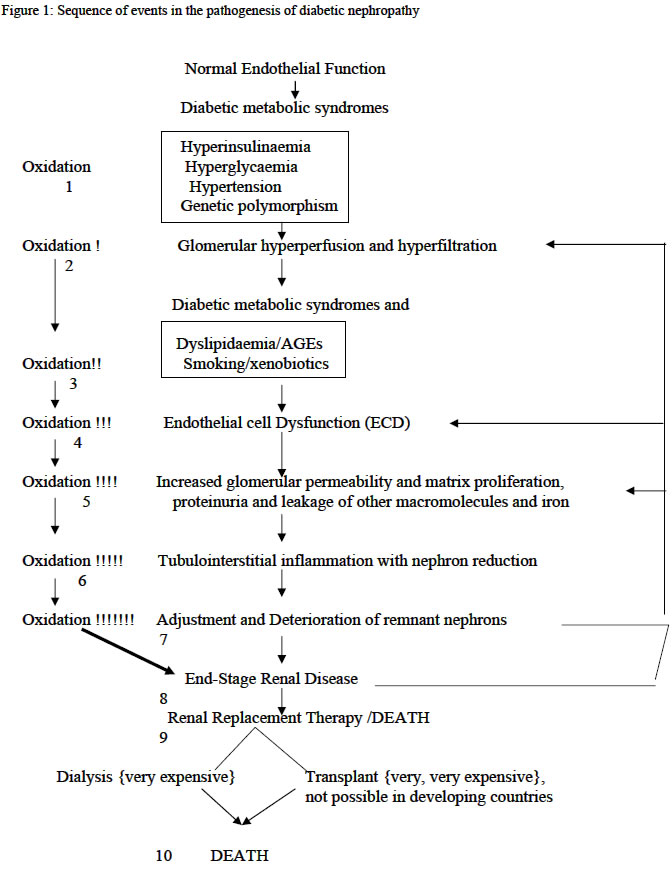
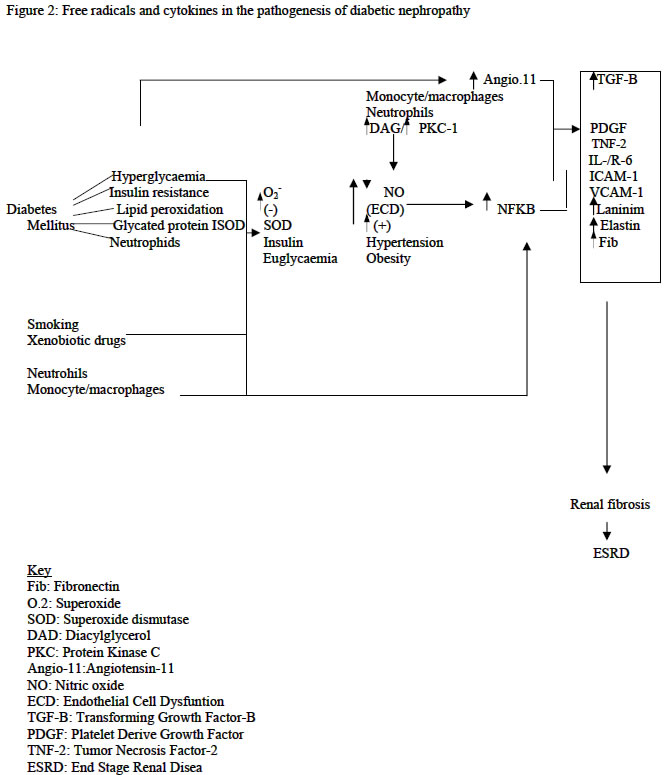
Hospital, Maiduguri, Nigeria Code Number: am04015 ABSTRACTDiabetes mellitus has assumed epidemic proportions in most parts of the world including the developing countries, and one of its ominous complications, diabetes nephropathy represent today the leading cause of end-stage renal disease in the developed countries. However, the pathogenesis of diabetes nephropathy remains illusive, notwithstanding, free radicals seem to be the most favorable linkage between all the associated factors suggested. Consequently, free radicals, oxidative stress and antioxidants have become commonly used terms in modern discussions of renal disease mechanisms, making the kidney unique among other organs as the site in which a spectrum of seemingly unrelated diseases involves reactive oxygen species. Importantly, hyperglycaemia and its attendant metabolic syndromes, smoking and the use of xenobiotics have been shown to accelerate free radical generations and attenuate the antioxidant system creating oxidative stress. The management of diabetes nephropathy is extremely expensive and frustrating. Therefore, prevention is better. Sources of antioxidants, especially antioxidant vitamins are available and affordable in most environments. These may be adjunct to other ways of preventing the development of diabetic nephropathy. Reviews like this are necessary to stimulate stakeholders in management of diabetes mellitus and modern nephrologists. Key words: Diabetes mellitus, nephropathy, free radicals, pathogenesis INTRODUCTIONMcCord and Fridovich opened the field of free radicals and oxidative stress when they discovered an enzyme superoxide dismutase (SOD). 1 Since then reactive oxygen species (ROS) has come to occupy an amazingly central role in a wide variety of diseases. Consequently, free radical, oxidative stress, and antioxidants have become commonly used terms in modern discussions of renal disease mechanisms, making the kidney unique among other organs as the site in which a spectrum of seemingly unrelated diseases involves ROS. 2 - 5 Diabetes mellitus and one of its complications, diabetic nephropathy, represent a leading cause of end-stage renal diseases (ESRD) in the developed countries especially United States and Europe. 6, 7 Hyperglycaemia of diabetes mellitus and its attendant metabolic syndromes 8 - insulin resistance, hyperglycaemia, hypertension, dyslipidaemia, obesity, and some social characters of these patients e.g. smoking and the use of xenobiotics, predispose to diabetic nephropathy. These accelerate free radical generation and attenuate the antioxidant defense system creating oxidative stress 2, 9 - 13.Consequently, increased free radical production and attenuation of antioxidant system is currently receiving the highest attention when discussing diabetic nephropathy. Therefore, stakeholders in management of diabetes mellitus as well as modern nephrologists are recommended to eliminate or prevent the development of the sources of free radicals right from diagnosis of diabetes or even in those with impaired glucose tolerance. PATHOGENESIS OF DIABETIC NEPHROPATHY The pathophysiology of diabetic nephropathy can be viewed as a sequence of events evolving in a stepwise pattern as shown in figure 1 and 2, where it starts with endothelial cell dysfunction (ECD) and ends with end-stage renal failure. However, ECD is preceded by glomerular hyperperfusion and hyperfiltration. The normal
endothelial cell Hence, NO production or availability can regulate diverse functions in endothelial cells. Therefore, any event that alters the function of NO will lead to endothelial cell dysfunction (ECD). Endothelial cell dysfunction
(ECD) What triggers ECD in diabetes
mellitus? a) High glucose can directly cause ECD and increases oxidative stress in glomerular mesangial cells, a target cell of diabetic nephropathy. b) Lipid peroxides and 8-hydroxydeoyguanosine, indices of oxidative tissue injury, were increased in the kidneys of diabetic rats with albuminuria. c) Oxidative stress induces mRNA expression of NFkB genes which in turn promotes production of proinflammatory proteins-TGF-B, fibronection, laminin, elastin, IL-1, IL-6, and PDGF, and d) Inhibition of oxidative stress ameliorates all the manifestation associated with ECD and diabetic nephropathy. Diabetic nephropathy is preceded by glomerular hypererfusion and hyper filtration, 7, 20 –22 which occur early in type 1 and in some 15-44% of type 2 diabetic patients at diagnosis, and these play a pathogenetic role in ECD, the early stage of diabetic nephropathy. 7, 20, 21 The hyperinsulinaemia of diabetes mellitus cause increase reabsorption of glucose and sodium in the proximal convoluted tubule and synthesis of NO via activation of IRS-1, 2. The insulin resistance also diverts more glucose into the pentose phosphate pathway, resulting in increase activation of PKC pathway and increase glomerular prostaglandins production, afferent arteriolar dilatation and efferent arteriolar constriction. 7, 20 - 23 Hyperglycaemia depresses the synthesis of heparin sulphate proteoglycans with consequent impairment of electrostatic barrier. 20These factors associated with genetic factors, e.g., genetic polymorphism of ACE and the sodium/lithium counter transport gene, lead to glomerular hyperperfusion and hyperfiltration. 23 The high GFR (about 20-30% increase) in humans has been shown to be strongly associated with nephromegaly. 21 These factors do not induce such early glomerular events alone but in tandem with oxidative changes. 7, 20 - 23 There is little doubt that the primary causal factor for the development of most diabetic complications is prolonged exposure to hyperglycaemia. The mechanisms of damage involve, 20, 23, 25 onenzymatic glycation of proteins, flux of glucose through the sorbitol pathway, increased de novo synthesis of diacylglycerol and subsequent activation of the protein kinase C pathway, impairment of electrostatic barriers, and increased lipid peroxidation. These not only generate ROS but also attenuate antioxidant mechanisms creating a state of oxidative stress. The process of nonenzymatic reaction of glucose with proteins is well known phenomenon and has been proven to account for numerous features of chronic diabetic complications. Advance glycosylation end products (AGEs)- receptor specific interactions with endothelial cells and macrophages, binding to antioxidant enzymes, and lipoproteins can lead to widespread ECD. 6, 20, 26, 27 Lipoprotein trapping and oxidation, deposition of AGEs in glomerular wall, inflammatory cell recruitment and activation, enhanced by clotting abnormalities and platelet activation, follow this. All these events augment production of free radicals. 6, 26, 27 There exist in diabetes alteration of blood pressure regulating system especially vascular reactivity as well as volume/sodium homeostasis. 28, 29 Adherence of diabetic red blood cells to endothelium results in an increase in production of lipid peroxides, and the resulting oxidant stress leads to a marked increase in PECAM-1 phosphorylations and transmigration of monocytic cells. Hyperglycaemia also promotes leukocytes adhesion to the endothelium through up regulation of cell-surface expression of adhesive proteins, and these processes depend on NF-Kb activation. These factors are part of the abnormal blood pressure deregulations. The glomerular endothelium experiences three primary mechanical forces, 32 the transmural pressure, the circumferential stretch (tension) and the shear stress. The altered blood pressure regulation/hypertension alters the shear stress. This appears to result in altered Intercellular Adhesion Molecule (ICAM-1), Vascular Cell Adhesion Molecule (VCAM-1) expression and increase monocyte adhesions, suggesting that the endothelial cells exposed to chronic arterial hypertension may lead to progressive ECD and disrupt the filtration barrier. 28 This indicates that oxidative stress is eminent in diabetic hypertensive patients causing ECD and therefore diabetic nephropathy. Abnormalities in lipid metabolism and the potential nephrotocixity have been suggested more than a century ago, 30, 31 and these are evidence by. 32 High cholesterol diets are associated with albuminuria and glomerular injury, lipid lowering agents and intake of large amount of dietary omega-n3 and –n6 reduced glomerular injury. Diabetes mellitus and renal tissue in this milieu has been suggested to accelerate oxidation of lipids. 6, 20, 23, 26. Diabetes mellitus as an oxidant milieu enhances free radical generation via; 6, 19, 33, 34 (a) non-enzymatic glycosylation of proteins (apolipoproteins, enzymes and receptors) leads to chronically elevated lipids and lipoproteins, (b) tincrease glucose metabolism through the aldose reductase pathways altering the intracellular redox balance, increase de novo synthesis of diacylglycerol and subsequent activation of PKC pathway, and the attenuation of antioxidant mechanisms create an oxidative stress. These predispose the chronically elevated lipids and lipoproteins to free radical oxidation. It has also been found in diabetic patients that lipid peroxides and 8-hydroxydeoxyguanosine, indexes of oxidative tissue injury were increased in diabetic patients. The oxidized lipids are toxic to tissues especially the vascular endothelium, glomerular mesangial cells, smooth muscle cells and renal tubular epithelium. Under this milieu the major cells of the glomerular wall can carry out lipid oxidation. 2 - 4, 20, 25, 26 Oxidized LDL exerts many biological effects that may contribute to the ECD and progression of diabetic nephropathy (table 1). 11, 32, 34, 35 Although the above considerable evidence support the notion that lipid abnormalities contribute to renal injury in animal models of endogenous and diet-induced hyperlipidaemia, not all models of hyperlpidaemia develop renal injury. 30This apparent paradoxical relationship between hypercholesterolaemia, atherosclerosis and glomerular injury in these models could be the result of differences in types of plasma lipoprotein. 30 Obesity, diabetes mellitus, dyslipidaemia and hypertension are common, interrelated medical problems, and are associated with an increased risk of ECD, vascular as well as glomerular diseases. Cigarette smoking is shown to impose oxidative stress to the body generally, and this may enhance progression of ECD and therefore diabetic nephropathy. 13 The use of xenobiotic drugs by diabetic patients may also contribute to the development oxidative stress in the body and the kidney in particular. 13 Table 1: Oxidized lipids in ECD
Tubulointerstitial
nephritis In the setting of tubulointerstitial disease, oxidative stress may directly originate from interstitial inflammatory cells, from pathologic events arising from or related to glomerular damage, or from metabolic adaptations occurring in the surviving nephrons. 5,43,44 Glomerular disease in diabetes allows leakage of red blood cells, a good source of iron, into the urinary space. Consequently human conditions associated with clinical proteinuria particularly diabetic nephropathy, iron excretion in urine is elevated. 34 Iron catalyzes oxidative reaction on the epithelial surface or intracellularly via Fenton reaction, producing highly damaging hydroxyl radicals. 13 The metabolic burden imposed on remnant nephrons obligates increased oxygen consumption in surviving nephrons, and incurs increased generation of ROS. 45,46 Neutrophils and activated macrophages are rich source of ROS. These ROS produced under different mechanisms activate NFkB genes with increased production of NFkB gene-dependent cytokines, which independently or synergistically lead to messangial cell proliferations, extracellular matrix expansion, and renal mass fibrosis. The ESRD also is good source of free radical generation. It has been found that atherosclerosis is common in patients with end-stage renal disease. This is not merely reflects the high prevalence of the traditional cardiovascular risk factors such diabetes and hypertension but an independent effect of ESRD on the vasculature. 47 The association of ROS and atherosclerosis is a well-known phenomenon. ESRD accelerates generation of free radical via the metabolic burden imposed on remnant nephrons obligating increase oxygen consumption, the dyslipidaemia, hyperhomocycteinaemia, and hypertension, which are commonly associated with the ESRD. This explains the vicious cycle demonstrated in figure 1. There is therefore a gradual and chronic effacement of nephrons with consequent deterioration of renal functions culminating into renal failure. Therefore, the role of free radicals in the pathogenesis of ECD and hence diabetic nephropathy are as summarized in table 2. Table 2: Free radicals in ECD and diabetic nephropathy 5,9-11,36
Consequently, oxidative stress is surely an inevitable accompaniment of diabetic nephropathy (see fig. 1 and 2 and tables 1 and 2). Therefore, how can diabetic nephropathy-related oxidative stress be dealt with? Eating a diet rich in fruits and vegetables will ensue adequate levels of antioxidant nutrients in the tissue and help the body to resists disease-related oxidative stress. 13 Fruits and vegetables are available and affordable even in recourse-poor communities. CONCLUSION An understanding of the homeostatic function of the glomerular endothelium is important for the modern nephrologist. The role of nitric oxide in mediating many of the regulatory properties of the endothelium is now recognized, as well as a growing understanding of how excess free radicals, generated under hyperglycaemia and its associated metabolic syndromes considered to be risk factors for diabetic nephropathy, cause endothelial cell dysfunction with loss of nitric oxide bioavailability. Because many antioxidant therapies appear capable of improving endothelial cell function and the fact that administration of L-arginine has restored different experimental nephropathies, including experimental diabetic nephropathy, causal relationship between improved endothelial cell function and diabetic nephropathy can be investigated in humans. REFERENCES
Copyright 2004 - Annals of African Medicine
The following images related to this document are available:Photo images[am04015f2.jpg] [am04015f1.jpg] |
| |||||||||

{kind=link}
{kind=link}