 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
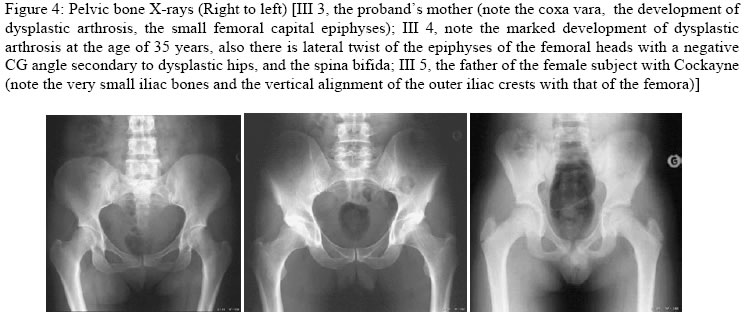
Annals of African Medicine, Vol. 4, No. 2, June, 2005, pp. 83-87 CONGENITAL DYSPLASTIC HIPS, SPINAL COLUMN ABNORMALITIES, FRACTURES AND PROGRESSIVE NEUROLOGICAL MANIFESTATIONS IN TUNISIAN FAMILY WITH COCKAYNE SYNDROME 1A. A. Kaissi, 1H. Safi, 1M. B. Ghachem, 2L. Hendaoui, 3F. B. Chehida 1Department of Paediatric Orthopaedics,
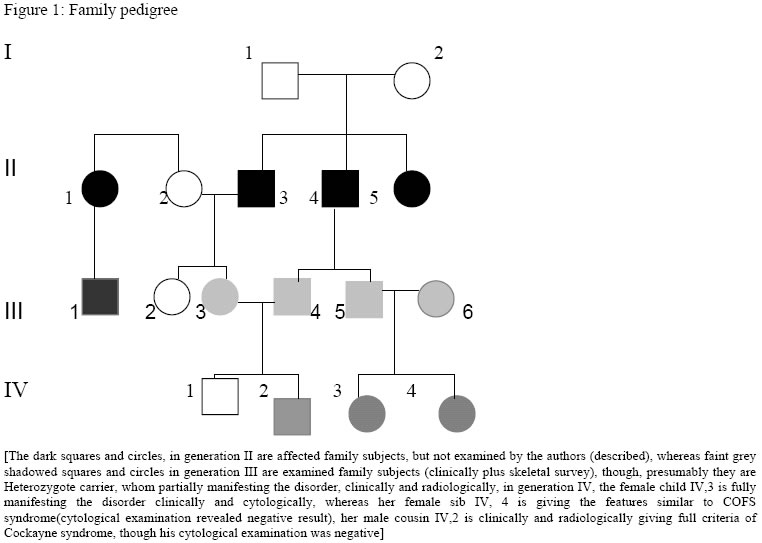
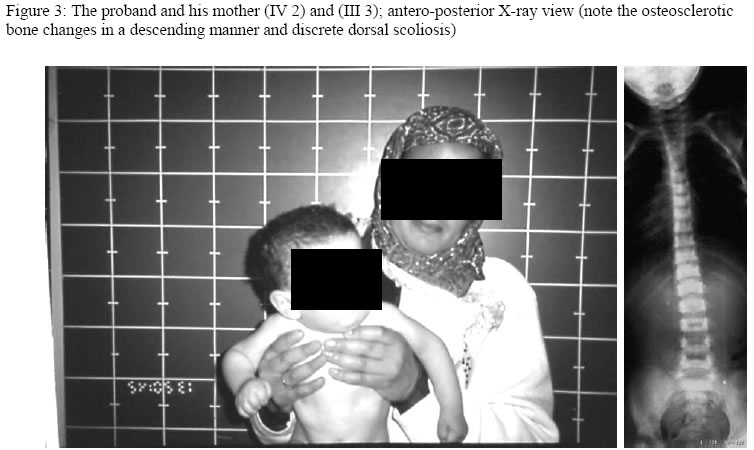
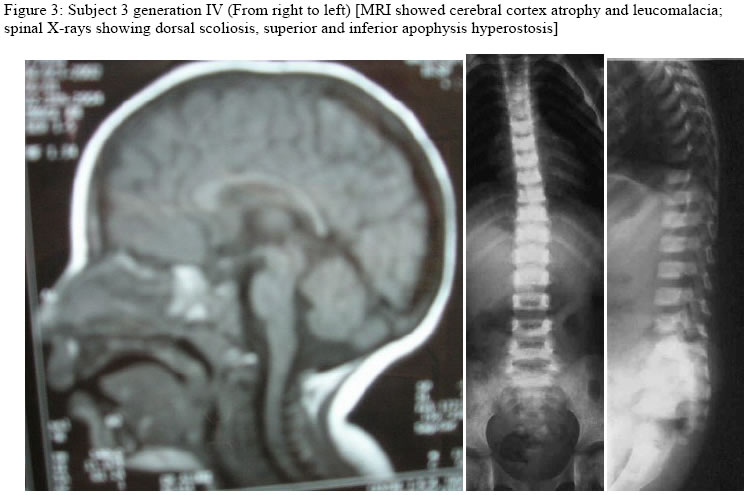
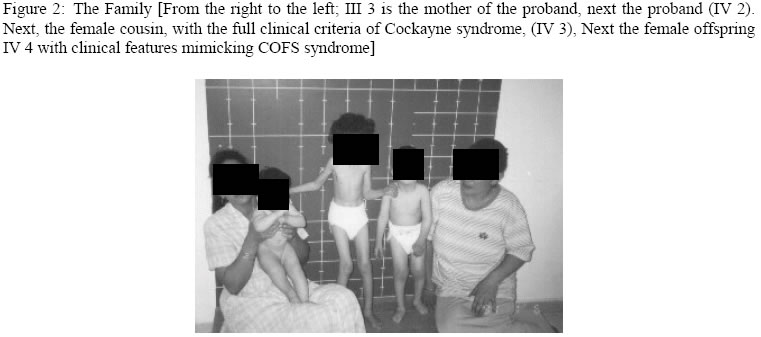
Hopital d’Enfants, Tunis, 2Department of Radiology, Hopital Monge Slim and 3Centre de Radiologie Ibn Zohr, Tunis, Tunisia Code Number: am05020 *A correction to this article has been published in Annals of African Medicine, Vol. 4, No. 3, 2005, pp. 141. Please see the related document for more details* Abstract We report an inbred, Tunisian family in which cousins have the definite diagnosis of Cockayne syndrome. Intervening members in this family, who are intellectually normal, though, most are manifesting complications of hip dysplasia (development of dysplastic arthrosis) and various vertebral abnormalities. We presume that these are carriers who manifest dreadful bone features rather than the clinical phenotype of Cockayne syndrome, the mode of inheritance of the abnormal gene in this family is suggesting autosomal dominant, to our knowledge the family reported with such skeletal abnormalities in association to Cockayne syndrome is the largest in comparison to the international literatures. Key words:Cockayne syndrome, skeletal abnormalities Résumé Nous faisons un rapport sur un cas résultant de croisements entre animaux de même souche, une famille tunisiene chez laquelle les cousins avaient un diagnostic précis du syndrome de cockaye. Les membres de cette famille qui interviennent et qui sont sains intellectuellement bien que la plupart des patients manifestaient des complications de la hanche dysplasie (devéloppement d’arthrose dysplastique) et des anomalies vertébrales. Nous supposons qu’elles sont des porteuses qui manifestent des traits épouvantables d’os plutôt que le phénotype clinique de syndrone de cockaye, la méthode d’héritage de ce gêne anomalie chez cette famille pourrait être autosome dominant. Pour autant que nous sachons, la famille s’est présentée atteinte d’une telle anomalité squelettique en association avec le syndrome de cockaye est le plus grand par rapport à la littérature internationale. Introduction In its classical form, Cockayne syndrome is a progressive neurological disorder characterized in infancy by sun-sensitivity, resulting in bullae and desquamation of the skin. The characteristic facial appearance does not develop until between the 2nd and 4th years of life when there is a loss of subcutaneous tissue around the eyes, giving the appearance of sunken eyes, as seen in premature ageing. The head circumference at this stage is small, as is length, and a sensor neural hearing loss is common. Both central and peripheral demyelination results in loss of skills, and features of a neuropathy, although limb reflexes (at the knee especially) can be exaggerated. A retinopathy occurs late and may be accompanied by optic atrophy. Pericapillary calcification in the cortex and in the basal ganglia is a common feature. Nance and Berry provide an excellent review. 1 We report a family (Figure 1), in which some gene carriers (heterozygotes) have facial features similar to those seen in homozygotes, and we assume that theseare manifesting carriers, a previously unreported finding in Cockayne syndrome, moreover in this family we encountered very specific bonechanges. Case reports The proband (IV 2-figure 3) was seen at the age of 7 days, referred to the department of paediatric orthopaedics because of congenital dislocation of the hips, and congenital contractures at the knees, elbows, and ankles. In the letter of referral the diagnosis was that of suspected cerebral palsy. The child was born, full term, and the birth weight, head circumference and length were all around the 10th percentile. The mother was a 32 year-old gravida 2, abortus 0, married to a 39-year-old first cousin. Her first pregnancy resulted in a male child with normal development. Examination of the proband revealed multiple congenital contractures, and marked hypertonicity in all 4 limbs. The Craniofacial examination showed sunken eyes due to loss or periorbital fat. The hands were small, and there was congenital dislocation of the hips, congenital scoliosis and pes cavus. Examination of the mother (III 3-figure 2) showed her height to be 146cm, her head circumference to be 53 cm. She was of normal facial appearance. Recently she begun experiencing pain in her pelvis and her hip joints, which she said, were stiffening progressively. Radiographs showed mild coxa vara and very small capital femoral epiphyses. The acetabular roof was abbreviated and did not adequately cover the capital femoral epiphyses. The father (III 4) had a height of 168cm and an OFC of 55cm. He was enophthalmic and had a cachectic face due to lack of subcutaneous fat. His hands and feet were large and radiographs of his pelvic bones showed spina bifida, and small capital femoral epiphyses. He had a dorsal scoliosis. The proband’s female cousin (IV 3-figure 4) who is 5 years old, is mentally handicapped. She was born at full term to first cousin parents and her weight, length, and head circumference were all around the 10th percentile. Gestation had been difficult, since her mother had experienced frequent bouts of heavy bleeding in the first trimester. This was treated with bed rest and weekly injections of primolout depot for 6 weeks, to prevent miscarriage. In the following trimester the fetal uterine movements were feeble and the child was hypotonic at birth and was diagnosed at the age of 6 months as having cerebral palsy. Seven months later torticollis was noted and examination revealed a unilateral congenital cataract, which was operated on at the age of 9 months. Radiographs showed vertebral flattening with superior and inferior Hyperostosis, and small pelvic bones. Her face was cachectic, with enophthalmos and lack of subcutaneous facial fat. She had long thin limbs, multiple contractures and stiffness of the joints. She IV 4 (figure 2) is the second female baby, was born full term, at birth she was hypotonic, and Microcephalic, and also was diagnosed as having cerebro-ocular-facial-skeletal (COFS) syndrome, when examined at our department at age of six months she showed, microcephaly, cataracts and joint contractures. The facial appearance is characteristic, in that the nasal root is prominent and the forehead slopes sharply backwards. Both the jaw and the eyes are small. There is good evidence that some infants diagnosed initially as COFS subsequently develop Cockayne syndrome including the sunken eye appearance, sensor neural deafness, photosensitivity, associated with marked developmental retardation. [The dark squares and circles, in generation II are affected family subjects, but not examined by the authors (described), whereas faint grey shadowed squares and circles in generation III are examined family subjects (clinically plus skeletal survey), though, presumably they are Heterozygote carrier, whom partially manifesting the disorder, clinically and radiologically, in generation IV, the female child IV,3 is fully manifesting the disorder clinically and cytologically, whereas her female sib IV, 4 is giving the features similar to COFS syndrome(cytological examination revealed negative result), her male cousin IV,2 is clinically and radiologically giving full criteria of Cockayne syndrome, though his cytological examination was negative] Table 1: Summary of subjects
Discussion The diagnosis of Cockayne syndrome depends on clinical and special investigations. The main clinical features are facial (sunken eyes) a progressive demyelinating neuropathy, dementia, retinitis pigmentosa and deafness. Brain showed cerebral cortex atrophy and leucomalacia. There is another clinical picture, as shown in the family presented here, in which the severity is greater and patients present with congenital joint contractures, mental retardation and cataracts. These patients proceed to develop the sunken eye appearance and at that stage the diagnosis of Cockayne becomes clearer. The interest of this family is that intervening members appear to be affected. Both fathers (who are brothers) of the proband and his cousin have deficient facial fat and seem prematurely aged. There have not been families with Cockayne who show dominant inheritance and given the cousin mating in this family there is little doubt that the mechanism of inheritance in this family is recessive. Heterozygote manifestation in recessive disorders is a rare phenomenon and carriers are usually totally normal. In this family there seems to be a multitude of manifesting carriers, and although the reason for this is not clear, this must be born in mind when assessing difficult pedigrees. This family also illustrates the variability of genetic conditions. This is more common in dominant inheritance where the locus on the unaffected chromosome seems to influence expression, but it is rarer in recessive inheritance where both loci are involved. Whether other genes on the same or a different chromosome affects expression and therefore clinical presentation, or whether environmental effects are involved, is unclear. One of the cousins presented with a hypertonic cerebral palsy-like picture, whereas the other was floppy with multiple joint contractures and a unilateral cataract. The condition in this family has been proven to be Cockayne syndrome by specialized tests (Chromosome breakage was seen on exposure of cells to UV light, in the female cousin- IV 3 in the family pedigree, whom manifested the full clinical plus the cytological criteria of Cockayne syndrome, whereas her female sib and her male cousin showed negative results to the chromosome breakage on exposure to UV light, this can explain the variability of the clinical and the cytological criteria of the disorder among the affected children in the same family, nevertheless, the only constant common features in this family were the radiological findings, moreover, looking at the clinical picture of the face, the sunken eyes especially, these occur very rarely in other conditions and together with the joint contractures and the cataract the diagnosis is compelling. The platyspondyly, and particularly the changes at the hips are well documented to occur in Cockayne syndrome, 2 - 9 although the striking findings in this family is the bone manifestations are of common occurrences in heterozygotes and of same intensity like these observed in the homozygotes. References
Copyright 2005 - Annals of African Medicine
The following images related to this document are available:Photo images[am05020f3a.jpg] [am05020f2.jpg] [am05020f3.jpg] [am05020f1.jpg] [am05020f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}