 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
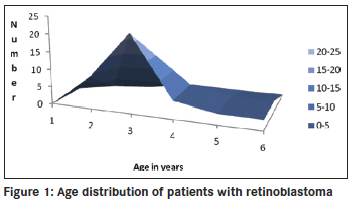
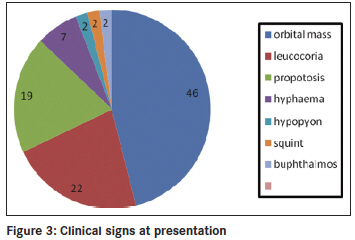
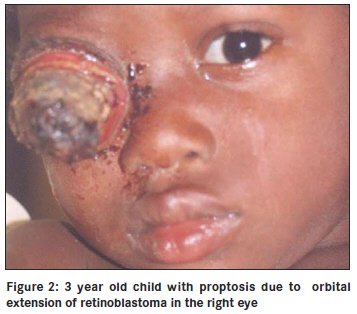
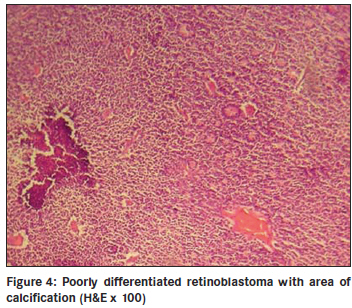
Annals of African Medicine, Vol. 10, No. 3, July-September, 2011, pp. 214-219 Original Article Clinicopathological pattern and management of retinoblastoma in Kano, Nigeria Lawan Abdu1, Sani Malami2 1 Department of Ophthalmology, Faculty of Medicine, Bayero University Kano, Nigeria Code Number: am11044 PMID: 21912005 Abstract Background: The aim of the study is to evaluate the pattern of presentation and the mode of management of retinoblastoma seen in Aminu Kano Teaching Hospital, Kano from 2001 to 2005. Keywords: Management, presentation, retinoblastoma Introduction Retinoblastoma is the commonest ocular malignancy in childhood. [1] The incidence is 1 in 20000 live birth in developed countries [2] and is likely to be higher in our environment. [3] The average age of diagnosis is 18 months and in vast majority of cases it present within the first 3 years of life. [1] Retinoblastoma can be familial or sporadic. In familial retinoblastoma inheritance is autosomal dominant with 90-95% penetrance and 6% of these have a positive family history and tend to be of early onset and bilateral. They tend to have second non ocular tumors. Patients with familial retinoblastoma have a 50% risk of transmitting the disease to their children. The genetic defect that predisposes to familial retinoblastoma is located on long arm of chromosome 13 (13q14). Sporadic cases may be unilateral or, bilateral. Bilateral cases are considered to have germinal mutations and are said to be gene carriers. Ten to 15% of unilateral cases are as a result of germinal mutation and carry the gene as do bilateral cases. [2],[4] This study highlights the pattern of presentation and clinicopathological features, and management of children with retinoblastoma over a 5 year period. Materials and Methods The clinic register was used to retrieve the case folders of all patients who presented with clinical features suggestive of retinoblastoma from 2001 to 2005. The following information was obtained; age, sex, tribe, and clinical details at presentation such as orbital mass, leucocoria, unexplained squint, hypopyon, endophthalmitis, uveitis in a child, or unexplained hyphema without history of trauma. The symptoms with duration at presentation were documented. Documented history of leucocoria was noted in patients who presented with orbital mass. Records of previous child with similar disease and history of application of traditional eye medicine were noted. All patients had basic eye examination. Vision was assessed in young children by fixation on light source, resistance to occlusion, and recognition of objects, picture charts, Landolts "C" chart or alphabet chart as the case may be. Basic eye examination included dilated fundoscopy of both eyes. Full clinical examination was conducted including examination of regional lymph nodes. Baseline investigations included full blood count (total and differential), hemoglobin genotype, urea and electrolytes, ECG, serum uric acid, orbital X-rays, and B scan was done for some of the patients. The patient′s weight was measured and recorded. The modality of treatment was either enucleation or modified exenteration. Specimen was sent for histopathology. For those who had enucleation a request was made of the histological pattern at the point of optic nerve section. Patients with tumor at the point of optic nerve section, or, orbital metastasis were placed on adjuvant chemotherapy. Results There were 42 patients between the ages of 2 to 6 years with retinoblastoma. There were 15 males and 27 females (M: F = 1.0:1.8). The youngest age at presentation was 2 years and the oldest child was 6 years old. The age distribution of the patients is shown in [Figure - 1] and [Figure - 3] years was the commonest age at presentation in 22 cases (55%). Forty patients were of the Hausas/Fulani tribe. Two other patients (5%) were of Ebira and Gwari ethnic background. There were 40 patients (95%) with unilateral tumor and 2 patients (5%) had bilateral disease. Tumor presented in the right eye in 20 patients (48%) and left eye in 22 patients (52%). One of the 2 patients with bilateral tumors was a product of consanguineous marriage. The parents have other children and none had the tumor. Overall there was positive history of first cousin marriages in 5 patients. None of the families had another child with retinoblastoma. There was positive history of application of traditional eye medication in 14 patients (33%) at a certain unspecified stage of the disease. The remaining 28 patients (67%) denied applying such medications. The interval between onset of symptoms and seeking medical assistance ranged between 4 weeks and 6 months. All the patients had been taken to at least two other health facilities before reaching our hospital. Parents of 29 patients (69%) admitted they suspected a malady about 4 months before going to any health center. The common presenting signs are shown in [Figure - 2] and [Figure - 3] with fungating orbital mass being the most common sign seen in 19 patients (46%). Those 15 patients (36%) who presented with leucocoria, hyphema, or hypopyon were requested to do a B scan. Nine patients (21%) did the scan and six could not afford the cost. B scan findings include retinal masses extending into the vitreous and with associated calcification in eight of the nine patients scanned. Twenty nine patients (69%) had anterior-posterior and lateral skull X rays taken, which showed no obvious calcification. The main radiographic appearance was soft tissue shadow in patients with orbital mass. The surgical management was modified exenteration in 31 patients (74%) and enucleation in 11 patients (26%). All specimens were sent for histopathology report. Forty-one patients (98%) had histologically confirmed retinoblastoma and 1 patient (2%) had toxocara granuloma. Of the 11 enucleated eyes, the optic nerve was tumor free at the point of section in three patients (7%) and five patients had tumor infiltrations at point of optic nerve section. While in three remaining patients of this group, the distal end of the optic nerve was not identified in the sections. Out of 41 patients with retinoblastoma, the tumor was well differentiated in 10 patients (24%), and poorly differentiated in 23 patients (55%) as shown in [Figure - 4]. Degree of differentiation was not specified in eight patients (19%). Of the 39 patients eligible for adjuvant chemotherapy, only 13 (33%) could be treated because of various reasons. The patients were to have six courses of the following agents: Vincristine 1.5 ng/m 2 , Cyclophosphamide 1000 ng/ m 2 , and Cytarabine 100 ng/m 2 . One patient with central nervous system involvement had intra thecal methotrexate. All the patients were placed on varied doses of Allupurinol daily during pulse therapy. Nine of the 13 patients had two courses before they missed subsequent appointment. Two patients requested for referral to another hospital closer to their home and only two had six courses of chemotherapy. Discussion Reliable population data on the incidence of retinoblastoma in our environment is not available, but hospital based series indicates that it is the commonest childhood ocular malignancy accounting for 60% of all childhood eye cancers. [5] It is claimed that retinoblastoma has a prenatal origin as occurrence is soon after birth [6] and there is a report of prenatal diagnosis of retinoblastoma [7] About half of our patients presented at the age of 3 years compared to 89% that presented before the age of 3 years in other parts of the world. [8] Retinoblastoma is also said to be commoner in African than Caucasian children, [9] and is characterized by late presentation in Africa. [10] The oldest patient in our study was 6 years old though there is a report of retinoblastoma in a 23 year old male, [11] and in the second (seeing eye) of a 27 year- old who had chemotherapy and Argon laser photocoagulation of the contra lateral eye in childhood indicating its rare occurrence in adults. [12] In our study, retinoblastoma was commoner in girls than boys though there is no sex predilection. Most of the patients in our study were of Hausa/Fulani tribe and this is probably due to the location of the hospital. There are intermarriages between and amongst the two groups such that many Hausas have mixed background. It is possible that unidentified environmental factors may play a role in rampant occurrence of the tumor in this group. The familial type of retinoblastoma is transmitted by autosomal dominant trait and consanguinity, a common practice among this group may not fully explain the frequent occurrence of the disease. None of the parents of the patients studied had a previous child with retinoblastoma. The risk of transmitting familial type of retinoblastoma is 50%. [2] Most of the patients presented months after symptoms were noticed, unlike the study in Ibadan where patients presented within one month of appearance of symptoms though only 5 out of 26 patients received full treatment. Many patients defaulted because of fear of enucleation and problems of treatment include financial constraints regarding investigations and medications, and availability of chemotherapy, [13] similar to the experience we had in our hospital. These factors contribute to late onset of treatment even for patients who presents with early disease. Socioeconomic and cultural reasons contribute to children presenting late with advanced disease in low income countries. The commonest mode of presentation was fungating orbital mass similar to the pattern in Nepal district of India where 40% presented with orbital mass. [14] Leucocoria was the second commonest presentation in one fifth of patients compared to 60% in other reports, another indication that patients present late. [15] We noticed that more than half of the parents noticed leucocoria at the early stage of the disease, but only a third had leucocoria at the time of presentation. With proper health education more parents (mothers) can detect this anomaly and report to the hospital early. Ultrasound B scan was requested for patients with leucocoria, not all the patients who needed to do the scan could afford the cost. B scan was helpful in making diagnosis particularly where the retinal view was obscured by secondary cataracts or, uveitis. High resolution B scan is helpful in early detection of retinoblastoma, particularly those in the anterior segment. [16] Our study showed that B scan is more useful in detection of intra ocular calcification than orbital X-rays, which showed no calcification in all the patients tested. Ocular ultrasound is useful in making a diagnosis and shows calcification in 91.3% in some reports. [17] Application of traditional eye medicine can worsen the symptoms complex leading to corneal melting or introducing infection. The goal of management is patient′s survival and globe preservation and treatment is tailored to each individual case. Various mode of management includes chemoreduction, focal therapy, laser photocoagulation, external beam irradiation, and crayotherapy. Radical treatment that results in enucleation is unnecessary in centers with better facilities. The international classification of retinoblastoma ICRB that grouped the tumor based on size and extent showed that most of our patients are in group E and present with extensive disease. This explain the reason for radical surgery in most cases as other alternatives such as chemotherapy with sub conjunctival Carboplatin proved to be successful elsewhere are unavailable. [18] Features of retinoblastoma invasion beyond lamina cribrosa in the optic nerve, choroid, sclera, orbit, or anterior chamber is an indication for chemotherapy. [19] Chemoreduction is a means of reducing tumor size with a view to apply more focal treatment, [20] and is an important step in the management of this disease. [21] Because the patients presented late, modified exenteration, which involved removal of all the orbital contents excluding orbital periosteum was the surgical operation performed in most of the patients, followed by enucleation. In developed countries, enucleation is reserved for cases with advanced disease with no hope for useful vision or when there is concern of invasion of neighboring structures. [22] Histologically, confirmed retinoblastoma was the commonest cause of removal of the eye in 48% of patients seen in our hospital. [23] Intra ocular tumor was the cause of removing the eye in 26% of children in Jos, Nigeria. [24] Despite the poor cosmetic effect, enucleation was the only treatment of choice in Lagos [25] for those without orbital extension. In Ibadan, Nigeria 37 patients (84%) had enucleation and 7 patients (16%) exenteration perhaps because the patients presented early. [26] Small tumors less than 3 mm in size in the posterior segment can be treated with focal laser photocoagulation. The aim is to apply two rows of laser burn around the tumor avoiding direct treatment as this can lead to vitreous seeding. [27] Crayotherapy is useful in treatment of small peripheral retinoblastoma. [28] External beam irradiation is known to offer good results in patients with advanced disease [29] as reported in our study though it is associated with risk of secondary tumors. [30] The clinical diagnosis of retinoblastoma correlated with the histological diagnosis in 98% of patients. Effectively, only a small number of patients without histological evidence of tumor invasion were cured. The tumor was poorly differentiated in more than half of the patients. Poorly differentiated tumors are commoner accounting for 58% in India [31] and 97% in Uganda. [32] Invasion of neighboring structures was commoner with undifferentiated retinoblastoma. [33] However, poorly differentiated retinoblastoma tend to be more radio sensitive. [34] We have no facilities for radiotherapy or crayotherapy in our hospital. In one patient the histology showed toxocara granuloma instead of the tumor diagnosed clinically. Intra ocular fine needle aspiration (FNAB) cytology under general anesthesia can be a viable option in making a diagnosis of retinoblastoma in selected cases. [35] Ratios of aqueous to plasma lactate dehydrogenase enzyme is high in eyes with retinoblastoma. [2] The technique is minimally invasive. Visualization of central retinal vessels with enhancement (contrast) on computed tomography (CT) scan indicates that the optic nerve is tumor free, [36] this is useful in making decision on mode of treatment. A patient on chemotherapy had central nervous system metastases and did not do well even with administration of intra-thecal methotrexate. Favorable response had been reported with methotrexate in other centers. [37] Neuroblastic intra cranial malignancy manifesting as intra cranial tumor is more associated with hereditary retinoblastoma. [38] Few of the patients who needed chemotherapy had a full course of the treatment. Many factors contributed to this, such as distance from the hospital, cost of multiple hospital admission, and cost of blood transfusion. Poverty is pivotal, as it cost about $45 (United States) per course of chemotherapy. In view of these factors, it is possible that many of the patients that were lost to follow up had eventually passed away without ever receiving full course of treatment. Decades ago mortality from retinoblastoma in Nigeria was 100%. [39] There is need to improve on early detection so as not to revert to the days of high mortality from this curable tumor. We are far away from 92% five year survival in Caucasian children. [40] Other modalities of treatment aimed at preserving vision such as chemo reduction, Argon laser photocoagulation, radio active plague therapy are better imagined in our environment. In our part of the world, the battle is to save lives first and then sight. It is therefore not surprising that the idea of use of anti cyclooxygenase 2 in years to come to treat retinoblastoma [41] may still be far from the reach of most of our patients. Our study indicates the limitations in access to facilities were children with retinoblastoma can be assessed early and treated promptly. There is cost attached to treatment and most of the families affected are of poor socioeconomic background. There is need to raise public awareness of this tumor and encourage funding of treatment by government and other support groups so that affected children are successfully cured. References
Copyright 2011 - Annals of African Medicine The following images related to this document are available:Photo images[am11044f4.jpg] [am11044f3.jpg] [am11044f1.jpg] [am11044f2.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}