 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Volume 6 Number 5, September/October 1996, pp.295- 300 Viral Load Quantitation:- An Integral part of Future Health Management.
Dianne Young, Roche Diagnostic Systems, Frenchs Forest, Sydney, NSW, 2086, Australia
Code Number: AU96016
Size of Files:
Text: 19.7K
Graphics: Line drawings (gif) - 42.2K
Our understanding of various viral infections has been rounded on the ability to accurately and precisely measure viral RNA in peripheral blood. The availability of reliable, standardised quantitative viral RNA assays with broad dynamic ranges has permitted the accumulation of a vast amount of information towards establishing prognosis and assessing the effectiveness of antiviral drugs in reducing viral load. In addition, the predictive value of this molecular information for subsequent clinical endpoints is now overwhelming. Consensus guidelines to consider in treating infected individuals using this molecular marker of infection have been drafted and have begun to appear in the literature. The baseline viral level as determined by Quantitative PCR, as well as the duration and magnitude of viral load changes, are parameters that merit closer scrutiny.
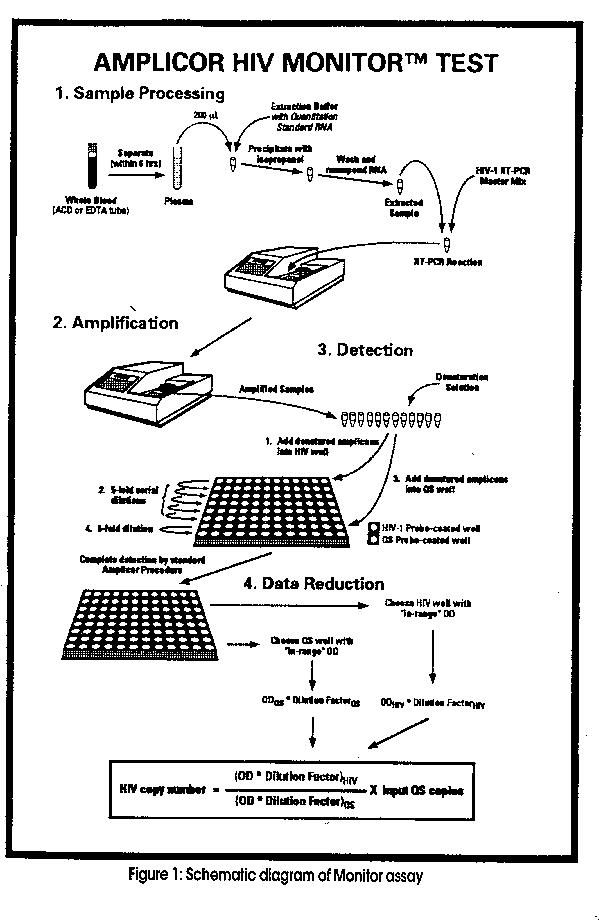
Nucleic Acid Amplification technologies are radically changing the nature of clinical diagnostics, providing new avenues for vast improvements in healthcare delivery. At the forefront of these technologies is the development of the polymerase chain reaction (PCR) assay. Since its introduction in 1984, PCR has caused a revolution in molecular biology research. But isotopic detection methods and the potential for carryover DNA contamination stood in the way of the introduction of the technique to the clinical laboratory environment. Today, however, standardised commercial kits featuring contamination control reagents, single use pre-packaged vials and non-isotopic detection methods, have paved the way for large-scale introduction of qualitative and, more recently, quantitative PCR into the clinical diagnostic laboratory. Standardisation A common assumption is that the exponential amplification of target nucleic acid sequences afforded by PCR might prevent if from being a quantitative procedure. However, there is now considerable evidence that under carefully controlled circumstances levels of PCR product correlate directly with the number of input molecules. There are various approaches to obtaining Quantification, with all requiring incorporation of some sort of 'Standard' in order to monitor variability in the assay that may be attributed to differences in reaction components, protocols or instrumentation. Critical decisions involve whether to include an 'Internal' standard (the design of which is described later) versus an 'External' standard (which entails plotting values obtained from a known concentration of the sequence to produce a standard curve). There is also the decision of whether to use an 'Endogenous' standard, (which is the co-amplification of a sequence that is already present in the sample, e.g. a HLA gene) versus an 'Exogenous' standard (addition of an additional target). Next is whether to use 'Competitive' versus 'Noncompetitive' amplification. In Competitive amplification, different levels of an internal standard are spiked into 5 to 7 tubes containing a constant amount of the target and coamplified. Measurement of the target is achieved by determining the concentration at which the internal standard and target signals are identical. The downside of this technique is that when two templates with similar sequences are co-amplified, the amplified product strands of the two templates may hybridize to each other and impede amplification. Non- competitive amplification can overcome this problem, as described later. Also to consider is 'Exponential' versus 'Plateau' amplification, where the efficiency of amplification is compromised due to various factors, such as limiting DNA polymerase concentrations or the annealing of plus and minus strands. Other factors affecting the quantitative nature of PCR include optimization of the reaction protocol (e.g. primer & nucleotide concentrations), avoidance of excessive cycling, adequate processing of samples to eliminate inhibitors that may decrease the efficiency of amplification, carryover prevention measures, selection of appropriate enzymes, not to mention choice of detection methods. As the use of PCR to quantitate RNA levels has already been extensively reported and reviewed elsewhere (Ferre 1992, Clementi et al 1993, Sninsky & Kwok 1993), this article will concentrate on the design of Roche Diagnostic Systems 'Quantitative RT-PCR Assay' format (AMPLICOR MONITOR TEST) which is currently used to quantitate HIV-1 and Hepatitis C virus RNA levels, with Hepatitis B virus and Cytomeglovirus in the near future. (See Fig. 1)
Sample preparation is straightforward and simply involves lysing viral positive plasma/serum samples with Guanidinium Isothiocyanate and subsequent RNA precipitation with Iso-propanol. The RNA is then resuspended in a specimen diluent which optimises buffer conditions for amplification.
Reverse Transcription and Amplification At the heart of the Amplicor Quantitative assays is a combined reverse transcription-PCR (RT-PCR) reaction which uses only one set of primers, a singe enzyme and one set of optimised buffer conditions. (Myers and Gelland, 1991) Typical viral reverse transcriptases used for the RT step have optimum enzymatic activity between 37-42 C. The discovery of a recombinant DNA polymerase derived from the thermophilic eubacterium Thermus thermophilus (rTth pol) which possesses efficient reverse transcriptase (RT) activity as well as PCR amplification ability, allows combined RT and PCR in the one tube. This dual function has been a major breakthrough for RT-PCR assays, and is vital to our Amplicor range of assays. RT activity proceeds at 60"C for 30 minutes to generate complementary DNA (cDNA). After this time the reaction conditions, in conjunction with the balance of cDNA compared to RNA, activates the polymerase subunit of the enzyme. The RNA-cDNA sequences are denatured by heating at 95 C for 1 minute, followed by repeated rapid cycling between 90 C and 60 C which directs the successive PCR steps of strand denaturation, primer annealing and fresh DNA synthesis. Reaction conditions compatible with the rTth pol performing both reverse transcription and DNA amplification involves the use of a single optimised buffer containing Mn^2+ and bicine rather than Tris-HCl and Mg^2+. Further benefits from the use of a thermophilic reverse transcriptase is the increased specificity of primer binding and alleviation of many secondary structures present in the RNA template, thus decreasing premature termination by the reverse transcriptase. Also, once the sample is added to the RT-PCR reagent master mix, there is no need to reopen reaction tubes, thus eliminating the major contamination source so often found with nested PCR assays. It also allows for the use of the UNG contamination control system as all reactions are performed at temperatures greater than 60 C (see below). Carryover Prevention Measures As with all Amplicor assays, we incorporate uracil- N-glycosylase and dUTP into the reaction mixture to ensure that any carryover of DNA from previous reactions will not compromise quantitation (Longo et al, 1990). Inclusion of an Internal Control The Quantitation of viral RNA is performed using an internal "Quantitation Standard" (QS) which is a non-infectious in vitro transcribed RNA transcript which is incorporated into each individual specimen at a known copy number. Both the QS and the Target share identical primer binding sites to generate products of equivalent length and base composition. The difference lies in a scrambled intervening region in the QS sequence to that of the Target sequence. This variance in the interior region enables independent detection of each amplified product by separate probes. As the introduced QS is carried through the extraction procedure, contains shared primer binding regions and is equivalent in length and base composition to the target, it is amplified with the same efficiency as the target. Therefore, the internal QS detects and compensates for any effects of inhibition, amplification efficiency caused by sample interferents, variability in reaction conditions, or thermal cycling in order to allow the accurate quantitation of each specimen. Further to this, as the plus and minus strands of the template and the standard cannot hybridise due to sequence variability, the reaction is considered to be "non-competitive". Also, the relative ampiicon levels are examined in the "exponential" phase to provide quantitative measurements. Colorimetric Detection The amplicons formed during the PCR process are labeled using biotinylated primers. Detection is not by gel electrophoresis but by hybridisation to standard ELISA-type microtitre plates whose wells are coated with a DNA capture probe. Two types of microwells are used:- one coated with a probe to the target DNA and the other with a probe to the QS. Following hybridisation a colour change is reproduced by addition of an avidin-horseradish peroxidase (HRP) conjugate, followed by a tetramethylbenzidine (TMB) substrate. As the internal QS is co-amplified with the target RNA specimen, absolute quantities of target copies can be extrapolated from the ratio of the target signal to the signal generated by the internal QS at a known number of copies. It should be noted that the target RNA copies are determined by using dilutions of the denatured product that are within the linear range of the standard curve. Quantitation Calculations RNA levels in the test specimens are determined by comparing the absorbance of the specimen to the absorbance obtained for the QS. The optical density in each well of the microwell plate is proportional to the amount of target or QS ampiicon in the well, and the total optical density is proportional to the amount of target or QS RNA, respectively, input into each reverse transcription/PCR amplification reaction. Hence, the amount of target RNA in each specimen is calculated from the ratio of the total optical density for the target specific well to the total optical density for the QS specific well multiplied by the input number of QS RNA molecules, using the following equation.
Total Target signal(OD x dilution factor)
Target RNA copies/mL = ---------------------------------------
Total QS signal (OD x dilution factor)
x Input QS copies per reaction
Hence, within 6 hours the Amplicor Monitor assay allows
quantitation of RNA levels using a single amplification
reaction and a simple colorimetric detection.Of intense current interest is the application of PCR Quantitation in measuring RNA viral load levels in HIV-1 infected persons. The natural history and pathogenesis of HIV infection are linked closely to the replication of the virus in the body. The infection is characterised by a variable clinical course with the development of AIDS approximately 7 to 11 years after infection. Immunologic damage occurs progressively through out the infection beginning during the early asymptomatic stage of infection. Ideally markers used to follow HIV infection should allow timely assessment of antiretroviral therapy, predict the ultimate clinical efficacy of new treatments, bear a biologically plausible relation to the disease process, be present in all patients, change rapidly in response to effective therapy, be derived from readily obtainable clinical specimens, and correlate directly with eventual clinical outcome.
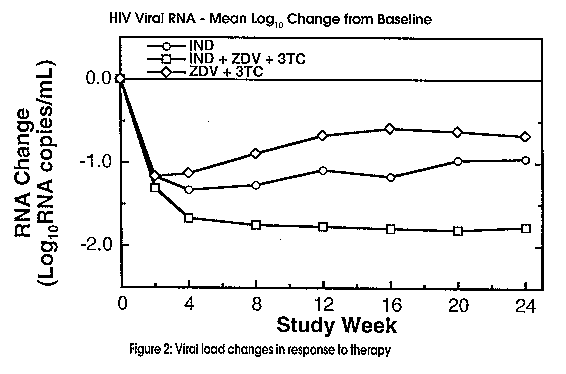
Most markers and studies to date have capitalized on the availability and relative non-invasive nature of peripheral blood samples even though the viral burden of an infected individual is distributed amoung multiple compartments (e.g. peripheral blood, lymph nodes etc.). The problem lies in that currently used surrogate markers (CD4+ cell count, p24Ag, beta2-microglobulin) have shown limitations in their ability to track the course of HIV infection. Recently, O'Brien et al, 1996 found that among a number of markers, changes in plasma HIV-1 RNA explained the effect of treatment on clinical outcome the most reliably and that changes in the CD4+ lymphocyte count provided additional information. He concluded that measuring viral load was an accurate means of monitoring progression of disease and provided an accurate assessment of a patient's response to treatment. His findings also suggested that viral load measurements would be a quicker way to obtain clinical endpoints in a trial as well as providing a more judicious use of expensive antiretrovirals. (Fig. 2)
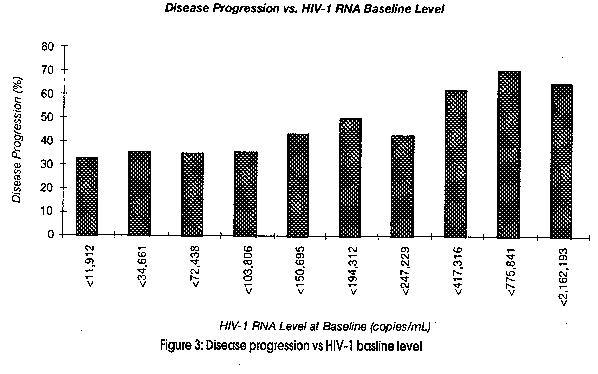
Accordingly, Mellors et al 1995 & 1996, showed that baseline levels of HIV-1 predicted clinical outcome following seroconversion, with a substantial increase in the rate of disease progression associated with higher baseline levels. (Figure 3)
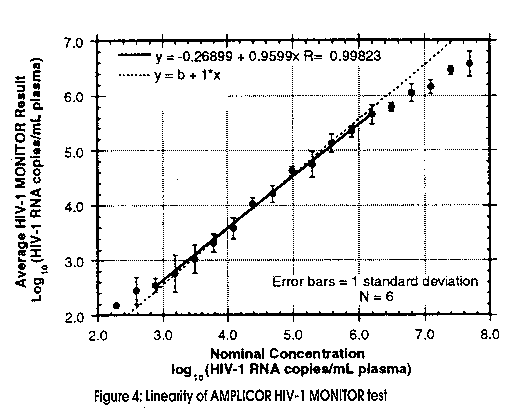
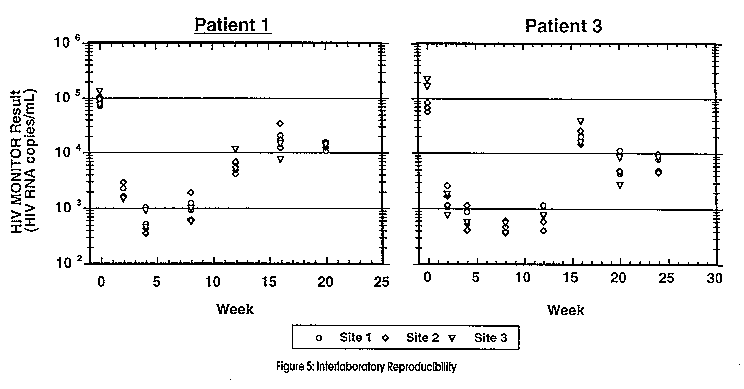
The challenge is therefore, to use HIV-RNA measurements to facilitate individual patient management decisions. For this reason, the precision, reproducibility, sensitivity and dynamic range of any Quantitative assay is of vital concern. In answer, Roche Diagnostic Systems has produced the AMPLICOR HIV-1 Monitor assay that currently reliably quantitates as low as 400 copies of RNA/ mL plasma (2 RNA strands per virus), with a prototype assay in the pipeline offering a sensitivity as low as 20 RNA copies/mL plasma. The upper detection limit of approx. 750,000 copies/mL can be increased further by diluting the sample in negative normal human plasma to bring it into the linear range. (Figure 4)
In summary, the development of assays that detect and quantitate HIV in clinical specimens, such as the Amplicor HIV-1 Monitor assay, has permitted insights into viral burden, replication dynamics, and antiviral drug resistance in infected patients. These insights will expedite the development of new drugs and the design of therapeutic strategies and will support the implementation of earlier and more aggressive antiviral therapy for HIV infection. Viral load quantitation will be an integral part of the health management decisions of the future. References Clementi, M., Menzo, S., Bagnarelli, P., Manzin, A., Valenza, A., Varaldo, P.E. (1993). Quantitative PCR and RT-PCR in Virology. PCR Methods Applic., 2. 191-196. Ferre, F. (1992). Quantitative or Semi-quantitative PCR: Reality versus Myth. PCR Methods Applic. 2. 1-9. Dickover R.E., Garratty E.M., Herman S.A., Sim M.S., Plaeger S., Boyer P.J., Keller M., Deveikis A., Stiehm R. & Bryson Y.J., (1996) Identification of Levels of Maternal HIV-1 RNA Associated with Risk of Perinatal Transmission. J.A.M.A., 275. 599-605. Longo, M.C., Beringer, M.S., Hartley, J.L. (1990). Use of Uracil DNA Glycosylase to Control Carry-over Contamination in Polymerase Chain Reaction. Gene. 93. 125-128. Mellors J.W., Kingsley L.A., Rinaldo C.R., Todd J.A., Hoo B.S., Kokka R.P. and Gupta P. (1995) Quantitation of HIV-1 RNA in Plasma Predicts Outcome after Seroconversion. Ann. Intern. Med., 122. 573-579. Mellors J.W., Rinaldo C.R., Gupta P., White R.M., Todd J.A. & Kingsley L.A. (1996). Prognosis in HIV-1 Infection Predicted by the Quantity of Virus in Plasma. Science, 272: 1167-1170. Mulder, J., McKinney, N., Christopherson, C., Sninsky, J., Greenfield, L., Kwok, S. (1994). Rapid and Simple PCR Assay for Quantitation of Human Immunodeficiency Virus Type 1 RNA in Plasma: Application to Acute Retroviral Infection. J. Clin. Micro. 32. 292-300. Myers, T.W. and Gelland, D.H. (1991). Reverse Transcription and DNA Amplification by a Thermus thermophilus DNA Polymerase. Biochemistry. 30. 7661-7665. O'Brien W.A., Hartigan P.M., Martin D., Esinhart J., Hill A., Benoit S., Rubin M., Simberkoff M.S., Hamilton J.D. (1996). Changes in Plasma HIV-1 RNA and CD4+ Lymphocyte Counts and the Risk of Progression to AIDS. N. Eng. J. Med. 334. 426-431. Sninsky, J., Kwok, S. (1993). The Application of Quantitative Polymerase Chain Reaction to Therapeutic Monitoring. AIDS. 7. (suppl 2) S29-S34. Copyright 1996 Australian Biotechnology Association Ltd. The following images related to this document are available:Line drawing images[au96016e.gif] [au96016c.gif] [au96016b.gif] [au96016d.gif] [au96016a.gif] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}