 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
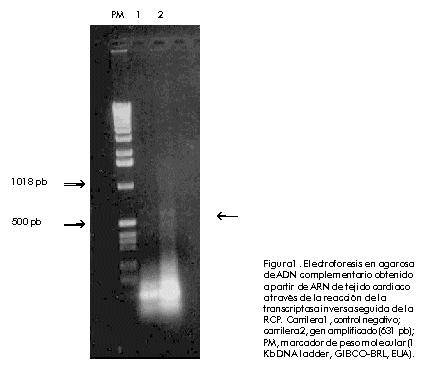
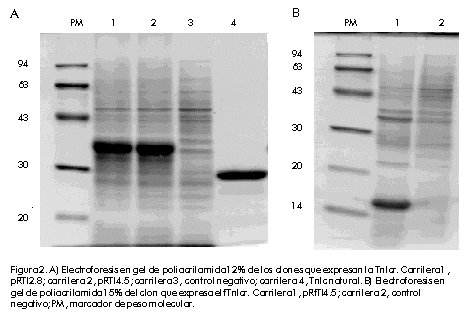
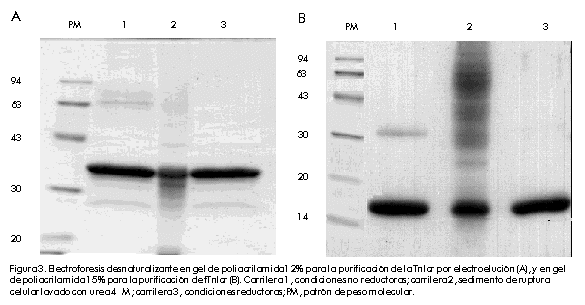
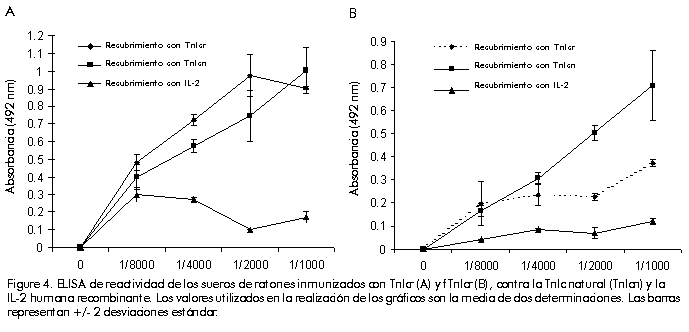
Biotecnologia Aplicada 2000; Vol. 17 No. 1, pp. 16-22 Clonación, expresión y purificación de la troponina I cardiaca humana y su fragmento aminoterminal en Escherichia coli Damián Mainet,1 Leticia Sánchez,1 Marcelo Nazábal,1 Maydel Echevarría,1 Luis Sorell1,2 1Centro de Ingeniería Genética y Biotecnología. AP 6162, CP 10600, Ciudad de La Habana, Cuba. Telf: (53-7) 21 8008; Fax: (53-7) 21 8070, 33 6008; E-mail: damian.mainet@cigb.edu.cu 2Instituto de Angiología y Cirugía Vascular. Calzada del Cerro 1551, Cerro, Ciudad de La Habana, Cuba. Recibido en febrero de 1999. Aprobado en agosto de 1999. Code Number: BA00005 RESUMEN En la actualidad, la troponina I cardiaca humana (TnIc) constituye el marcador bioquímico más cardioespecífico del infarto agudo del miocardio. La TnIc presenta una región aminoterminal de 32 aminoácidos adicionales no presentes en su isoforma esquelética, lo que le confiere cardioespecificidad. En este artículo, se describe el aislamiento y la amplificación del gen que codifica la TnIc, a través de la reacción en cadena de la polimerasa acoplada a la transcripción inversa del ARN total purificado a partir de músculo cardiaco. Posteriormente, se realizó la clonación del ADN amplificado en el vector de expresión. El fragmento aminoterminal cardioespecífico fue clonado de forma independiente en el mismo vector de expresión. En ambos casos, se obtuvieron niveles de expresión elevados de 43% y 30% en Escherichia coli, respectivamente. Estas moléculas se expresaron en su mayor parte como proteínas insolubles. Se purificaron por electroelución en geles de poliacrilamida y se obtuvieron rendimientos de 41 mg de TnIc recombinante y 23 mg del fragmento aminoterminal recombinante de la TnIc por litro de cultivo de E.coli. La pureza de ambas proteínas recombinantes varió entre 92% y 94%. Las dos variantes de las moléculas recombinantes, mostraron propiedades antigénicas similares a las de la TnIc natural mediante ultramicroELISA. Por esta razón, estas moléculas podrían constituir inmunógenos apropiados para obtener anticuerpos monoclonales antiTnIc cardioespecíficos, que se emplearían en el diagnóstico del infarto agudo del miocardio. Palabras claves: electroelución, expresión en bacteria, reacción en cadena de la polimerasa, troponina I cardiaca ABSTRACT Cloning, Expression and Purification of Human Cardiac Troponin I and Its Specific Amino-terminal Fragment in Escherichia coli. The human cardiac troponin I (TnIc) is nowadays the most cardiospecific diagnostic marker for acute myocardial infarction. The cardiac isoform of troponin I contains 32 additional amino acids on the N-terminus, which confer cardiac specificity. This paper describes the isolation of TnIc gen through reverse transcription-polymerase chain reaction using total RNA from cardiac tissue, and its cloning into an expression vector. The cardiospecific aminoterminal fragment of TnIc was cloned into the same expression vector. These molecules were obtained to high levels in Escherichiacoli, representing 43% and 30% of total cellular protein, respectively. Both molecules were highly expressed as insoluble proteins. The entire protein and its fragment were purified by electroelution with a 94% and 92% purity, respectively. The yields of recombinant cardiac troponin I and recombinant aminoterminal fragment of TnIc were 41 mgand 23mg per liter of E.coli culture, respectively. The recombinant TnIc and the fragment showed antigenic properties similar to those of the natural TnIc in ultramicroELISA. These molecules are thus good immunogens for obtaining anti-TnIc cardiospecific monoclonal antibodies, which are very important for the development of diagnostic kits for detecting acute myocardial infarction. Keywords: bacterial expression, cardiac Troponin I, electroelution, polymerase chain reaction Introducción Según la Organización Mundial de la Salud, los tres criterios fundamentales en el diagnóstico del infarto agudo del miocardio son: la historia clínica, el electrocardiograma y la determinación de enzimas séricas. La principal enzima utilizada para el diagnóstico de esta enfermedad es la creatina-quinasa (CK, EC 2.7.3.2) y su isoenzima MB (CKMB). Sin embargo, los niveles de esta proteína se elevan muchas veces en pacientes con trauma esquelético o que han realizado ejercicio muscular intenso [1]. Por esta razón, pierde su valor diagnóstico en esas situaciones. En la actualidad, se le presta atención a las proteínas estructurales y contráctiles del corazón para el diagnóstico del infarto agudo del miocardio. Esto se debe a la expresión de isoformas cardioespecíficas de estas macromoléculas, a su mayor concentración intracelular y a su continua liberación en el miocardio necrótico [2]. Las troponinas cardiacas T (TnTc) e I, que pertenecen a este grupo de proteínas, son más cardioespecíficas que la enzima CKMB. Además, por tener una ventana diagnóstica de 7 a 10 días con respecto a los 2 o 3 días de la CKMB, son de mayor valor en el diagnóstico tardío del infarto agudo del miocardio. Por otro lado, permiten valorar el pronóstico en pacientes con angina inestable, monitorear el daño miocárdico perioperatorio y la isquemia miocárdica en pacientes politraumatizados, y evaluar la terapia de reperfusión coronaria [3]. Estas macromóleculas ayudan a calcular el tamaño del infarto miocárdico [4,5]. Recientemente, se encontró que los niveles de la TnTc se elevan en pacientes con insuficiencia renal y rabdomiolisis, y su cardioespecificidad disminuye con respecto a la TnIc [6]. La TnI tiene un peso molecular de 24kD y ejerce un efecto inhibitorio en la actividad ATPasa, estimulada por Mg2+, de la actomiosina [7]. Existen tres isoformas de la TnI específicas de tejido en humanos: de músculo cardiaco, de músculo esquelético rápido y de músculo esquelético lento. La isoforma cardiaca contiene 32 aminoácidos (aa) adicionales en el extremo aminoterminal, los cuales le confieren especificidad cardiaca. Esta región aminoterminal contiene un residuo de serina cuya fosforilación forma parte del proceso de contracción muscular estimulado por agonistas b-adrenérgicos del corazón. El gen de la TnIc humana ha sido previamente clonado y expresado en E.coli como molécula fusionada o no fusionada a la b-galactosidasa. También, se expresó la porción N-terminal de ese gen fusionada al extremo 3' del gen de la b-galactosidasa. Los tres polipéptidos fueron reconocidos por un anticuerpo antiTnIc humano [8]. No se reportaron los rendimientos de la proteína fusionada TnIc-b-galactosidasa, ni del fragmento aminoterminal de la TnIc-b-galactosidasa. En 1995, Hayden et al. [9] clonaron y expresaron la TnIc humana como proteína heteróloga fusionada al extremo C-terminal de la CMP-KDO sintetasa (CTP: CMP-3-desoxi-d-mano-octulosonato-citidililtransferasa), enzima que está presente en bacterias gramnegativas. Los niveles de expresión de la proteína de fusión CMP-KDO sintetasa y TnI, correspondieron a 8% de las proteínas totales 4h después de la inducción con isopropil-b-d-tiogalactopiranósido (IPTG). Esta TnIc fusionada fue analizada mediante un ensayo inmunoenzimático de micropartículas y se encontró una respuesta antigénica similar a la de la TnIc aislada del tejido cardiaco humano. Este artículo describe la clonación y expresión de la TnIc y su fragmento aminoterminal cardioespecífico en el vector pR2M6 bajo el control del promotor triptófano, así como la purificación de ambos productos génicos. Se obtuvieron altos niveles de expresión con el uso de una secuencia estabilizadora de 58 aa que codifica la interleuquina-2 humana (IL-258) como segmento de fusión al extremo N-terminal de la proteína de interés. La purificación de estas proteínas por electroelución arrojó un grado de pureza mayor de 90%. Estas moléculas recombinantes constituyen una alternativa para la obtención de anticuerpos específicos contra la TnIc. Materiales y Métodos Reactivos Los oligonucleótidos fueron sintetizados en el Departamento de Oligonucleótidos del Centro de Ingeniería Genética y Biotecnología (CIGB, La Habana, Cuba). Las enzimas de restricción fueron adquiridas de Promega (Madison, EUA). El conjugado antiIgG de ratón-peroxidasa fue producido en la División de Inmunotecnología y Diagnóstico del CIGB. Los reactivos utilizados son de calidad para biología molecular y aquellos no especificados fueron comprados a la firma Merck (Alemania). La IL-2 humana recombinante, con un grado de pureza mayor de 95%, fue suministrada por el Departamento de Control de Calidad del CIGB. Extracción del ácido ribonucleico total La extracción del ácido ribonucleico (ARN) total se realizó por el método de un solo paso con tiocianato de guanidio-fenol-cloroformo, a partir de muestras de autopsia de músculo cardiaco humano. A dicho protocolo, descrito por Chomczynski y cols. [10], se le realizaron algunas modificaciones. A 1g de tejido cardiaco en hielo, se le añadieron 10mL de la solución D (tiocianato de guanidio 4M, citrato de sodio 25mM pH 7, 2-mercaptoetanol 0,1M), después se homogeneizó con politrón (Janke & Kunkel, Alemania) durante 2 min en hielo. Al tejido homogeneizado se le añadió Sarkosyl 0,5% (concentración final) y se agitó en vórtex (Janke & Kunkel, Alemania). Luego se centrifugó a 10900xg, a 4ºC por 10min. Al sobrenadante se le añadió 1mL de acetato de sodio 3MpH 5,2, 10mL de fenol saturado con agua y 2mL de una mezcla de cloroformo: alcohol isoamílico (49:1); se agitó vigorosamente por 2min, se incubó en hielo 15min y se centrifugó a 10900 xg, a 4ºC por 20min. Se descartaron las fases fenólica e intermedia, y a la fase acuosa se le añadió igual volumen de isopropanol, se incubó a -20ºC por 1h y se centrifugó a 10900 xg, a 4ºC por 20min. El precipitado se homogeneizó en 0,6mL de la solución D y se transfirió a viales de 1,5mL. Luego, a dicho homogenado se le añadieron 0,06mL de acetato de sodio 3M pH 5,2 y seguidamente se adicionaron 0,66mL de isopropanol, se incubó a -20ºC por 1h y se centrifugó a 10900xg, a 4ºC por 10min. El precipitado de ARN se resuspendió en etanol 75% y se centrifugó a 10900xg, a 4ºC por 10min. El precipitado se secó al vacío durante 15min y se resuspendió en 200µL de agua con dietilpirocarbonato. La concentración de ARN total se determinó por la fórmula: concentración de ARN (µg/mL) = DO260 x 40µg/mL x factor dilución [11] donde: DO: densidad óptica Para analizar la pureza del ARN total, se realizó una electroforesis en gel de agarosa 1% que contenía formaldehído 2,2M [11]. Síntesis del ácido desoxirribonucleico complementario y reacción en cadena de la polimerasa El ácido desoxirribonucleico complementario (ADNc) de la TnIc fue obtenido mediante transcripción inversa, seguido de la reacción en cadena de la polimerasa (RT-RCP), a partir de ARN extraído de corazón humano. Se diseñaron dos cebadores complementarios a los extremos 5' y 3' de la secuencia de la TnIc humana reportada en la base de datos EMBL [12]. El oligonucleótido del extremo 5' de la TnIc fue denominado tropo 5 e hibrida de la posición 1 a la 24 (5'-CTCTAGAAATGGCGGATGGGAGCAgcgatggc-3'), y el oligonucleótido del extremo 3' fue denominado tropo 3 e hibrida de la posición 600 a la 624 (5'-GCGTCGACTCTTGCGGCCCTCCATTCCACTCAG-3'). Se incluyeron los sitios XbaI y SalI para la clonación del ADNc en el vector de expresión. La síntesis del ADNc se realizó como sigue: se hibridaron 170 pmol de tropo 5 y 80pmol de tropo3 con 4mg de ARN de corazón humano en 33mL de agua destilada, 10µL del tampón 10X (Perkin Elmer, EUA), 0,2mM de cada uno de los desoxirribonucleótidos dATP, dCTP, dGTP y dTTP (Amersham, Reino Unido), 5mM de ditiotreitol, 5 U de transcriptasa inversa (Boehringer Mannheim, Alemania), 5 U de Taq ADN polimerasa (Amersham, Reino Unido), en un volumen final de 100µL. Se calentó la reacción a 94ºC por 10min y luego se incubó por 1h a 42ºC. La RCP consistió en 40 ciclos de desnaturalización a 94ºC por 1min, hibridación a 60ºC por 2min y extensión a 72ºC por 1min, seguido de un paso final de polimerización a 72ºC por 5min, en un termociclador Minicycler PTC-150 (MJ Research, Massachusetts, EUA). Clonación del gen de la TnIc y su fragmento aminoterminal en los vectores de expresión Los fragmentos de RCP fueron visualizados en geles de agarosa tipo I (Sigma, EUA) 0,8% teñidos con bromuro de etidio (Boehringer Mannheim, Alemania) según Maniatis y cols. [11]. Los productos de la RCP fueron purificados por el método de recobrado en gel de agarosa de bajo punto de fusión [11], fueron digeridos con las enzimas de restricción XbaI y SalI, y ligados al vector de expresión pR2M6 [13], previamente tratado con XbaI y SalI [11]. El fragmento aminoterminal de la TnIc fue obtenido a partir del clon pRTI4.5. El plasmidio fue digerido con las enzimas BamHI y BglII [11]. El fragmento del plasmidio que contiene la región aminoterminal de la TnIc humana, fue purificado por el método de recobrado en gel de agarosa de bajo punto de fusión y se ligó utilizando la ADN-ligasa del fago T4 [11]. Los clones que contenían el fragmento se seleccionaron según su talla en geles de agarosa 0,8% y luego se verificaron por análisis de restricción con la enzima FokI. El clon seleccionado para el estudio se denominó pRfTI4.5. Hibridación de colonias y secuenciación La cepa XL1-Blue de E. coli fue utilizada para la introducción por transformación del producto de la ligación [11]. Los transformantes positivos fueron seleccionados por la técnica de hibridación de colonias [11], con el empleo, como sonda, del oligonucleótido interno correspondiente a la región 509-539 de la secuencia de la TnIc humana (5'-GACCTGCGGG CCCACCTCAAGCAGGTGAAG-3'). El oligonucleótido fue marcado con 32P (Amersham, Reino Unido) mediante la reacción de la polinucleótido-quinasa, a partir de [g-32P]dATP (Promega, EUA) [11]. Los plasmidios aislados que contenían las secuencias de la TnIc, pRTI4.5 y pRTI2.8, fueron usados para la secuenciación parcial del gen. Dichos plasmidios fueron secuenciados por el método de Sanger y cols. [14], mediante el protocolo de los fabricantes del estuche de reactivos DNA fmolÒ (Promega, EUA). Para la secuenciación se utilizaron los oligonucleótidos tropo 5, uno interno que hidrida en la región 144-164 del gen de la TnIc humana (5'-AAATTGCAGCTGA AGACTCT-3') y uno que hibrida en la secuencia terminadora del fago T4 (5'-GGTCATTCAA AAGGTCATCCAC-3'). La región correspondiente al segmento de gen insertado en el plasmidio pRfTI4.5 fue secuenciado con el oligonucleótido tropo 5. Expresión de las proteínas de fusión IL-258-TnIc e IL-258-fTnIc Las células de la cepa XL1 Blue de E.coli fueron transformadas con el plasmidio pRfT14.5 y cultivadas en 3mL de medio Luria Bertani (LB, triptona 10mg/mL, extracto de levadura 5g/L, NaCl 10g/L, pH 7,4) suplido con ampicillina a una concentración final de 50µg/mL (LBA), por 12h a 37ºC con agitación. Dicho precultivo se inoculó en 50mL de medio mínimo M9 que contenía hidrolizado de caseína 1% y glucosa 0,4%, y se incubó a 37ºC con agitación por 6h. La expresión se indujo con la adición de ácido b-indolacrílico a una concentración final de 0,27mM, y la incubación del cultivo por 2h bajo las mismas condiciones. La expresión de la proteína se analizó a partir del sedimento de centrifugación del cultivo inducido, mediante electroforesis desnaturalizante en geles de poliacrilamida (SDS-PAGE) 12% para la TnIcr y 15% para el fTnIcr, según Laemmli [15]. El por ciento de expresión fue calculado por densitometría a partir de los geles de poliacrilamida teñidos con Coomassie blue G-250 (BioRad, EUA). Purificación de las proteínas de fusión IL-258-TnIc e IL-258-fTnIc La cepa XL1 Blue de E. coli transformada con el vector pRTI4.5, se inoculó en 50mL de medio LBA y se incubó toda la noche a 37ºC con agitación. Este cultivo fue inoculado en 500mL del mismo medio hasta una DO600 aproximada de 1,3. La expresión se indujo con la adición de ácido b-indolacrílico a una concentración final de 0,27mM, y con incubación durante 2h adicionales a 37ºC. Las células fueron cosechadas por centrifugación. Con el plasmidio pRfTI4.5 se procedió de manera similar, excepto que el precultivo fue inoculado en 300mL de LBA y las células se dejaron crecer hasta una DO600 de 0,8. Las células se lavaron con Tris-HCl 10mM pH8 y EDTA 1mM, luego se trataron con ultrasonido (sonicador iKa LabortechniK, Janke & Kunkel, Alemania) a una potencia de 74W/cm2 durante tres ciclos de 1min de tratamiento y 1min de incubación en hielo. La solución de células rotas se centrifugó a 2500xg durante 15min, se descartó el sobrenadante y el sedimento se resuspendió completamente en urea 4M. Se volvió a centrifugar y este procedimiento de lavado en urea 4M se repitió dos veces más. La electroelución fue el método empleado para la purificación de las proteínas recombinantes. Con este propósito, se realizó una SDS-PAGE a escala preparativa (gel de 13cm x 15cm x 0,3cm a 12,5%), en la que se aplicaron 60mg, en peso húmedo, del sedimento lavado disuelto en Tris-HCl pH 6,9, b-mercaptoetanol 0,1%, SDS 4% y glicerol 20%, y calentado a 100ºC en un bloque térmico (ISPJAE-tp, Cuba) durante 20 min. La corrida electroforética se hizo a una intensidad de corriente de 25mA. Luego de la tinción inversa del gel según el protocolo de Imidazol-ZnCl2 [16], se separó la banda correspondiente a la proteína recombinante. Este segmento del gel se pasó cinco veces a través del orificio de salida de una jeringuilla de 10mL. El gel se pasó a un tubo de diálisis que retiene proteínas mayores de 10kD (Spectrum, EUA), el cual contenía NH4HCO3 0,4M, SDS 0,2%, b-mercaptoetanol 0,1%, y se colocó en una cámara de electroforesis horizontal. El tampón de electroforesis contenía NH4HCO3 0,04M, SDS 0,2% y b-mercaptoetanol 0,1%, y la corrida electroforética se hizo a 50 V durante 16h. La proteína electroeluída se dializó contra tampón PBS (KH2PO4 0,2g/L, Na2HPO4 11,5g/L, KCl 0,2g/L, NaCl 0,8g/L, pH7,2) y se determinó su concentración por medición de la DO a 260 nm y 280nm, según la fórmula: concentración de proteína (mg/mL) = (DO280 x 1,55) - (DO260 x 0,76) [17]. El grado de pureza se calculó por análisis densitométrico del gel de poliacrilamida teñido con Coomassie blue G-250. Obtención del suero antitroponina Los ratones Balb/c fueron inmunizados por vía subcutánea con 20µg de TnIcr o fTnIcr en adyuvante completo de Freund 50%, y las otras tres dosis en adyuvante incompleto de Freund 50%. Se extrajeron 200µL de sangre por animal y se centrifugaron a 1000xg para obtener el suero, el cual se conservó a -20ºC hasta el momento de su uso. Análisis inmunoenzimático antiTnIcr o antifTnIcr Las placas de ultramicroELISA (Tecnosuma International S.A., La Habana, Cuba) fueron recubiertas con 20µL de soluciones de TnIc natural (Biodesign International, EUA), TnIcr o fTnIcr a una concentración de 1µg/mL, e IL-2 recombinante a 10µg/mL en tampón carbonato-bicarbonato de sodio 0,05M pH9,6 durante 12h a 4ºC. Los pozos se lavaron con una solución de Tween 20 0,05% preparada con agua desionizada. El lavado se repitió tres veces. Se aplicaron 25µL de la solución de bloqueo (PBS, leche deslipidizada 2% y Tween 20 0,05%, pH 7,2) y las placas se incubaron por 1h a 37ºC. Después se realizaron tres lavados. Se añadieron las mezclas de suero de ratones inmunizados con TnIcr o fTnIcr y la mezcla de sueros de ratones preinmunes (20µL por pozo) diluidos en tampón diluente (PBS, leche deslipidizada 1% y Tween 20 0,05%). Las placas fueron incubadas por 1h a 37ºC. Después de cinco lavados, se añadieron 20µL/pozo del conjugado antiIgG ratón-peroxidasa (1:4000 en el tampón diluente) y se incubó por 1h a temperatura ambiente. El anticuerpo antiIgG de ratón utilizado en el conjugado fue obtenido en carnero. Después del lavado, se desarrolló el color por incubación con 20µL de la solución de revelado durante 10min. La solución de revelado consistió en o-fenilendiamina 5mM disuelto en tampón citrato-fosfato de sodio 100 mM pH5,0 y H2O2 0,015% (v:v). La reacción se detuvo mediante la adición de 5µL de H2SO4 2,5M y la absorbancia se determinó a 492nm en un lector colorimétrico de microplacas de ELISA (Titertek Multiskan, Flow Laboratories, Finlandia). Resultados Análisis de la clonación y la expresión de TnIcr y fTnIcr La purificación del ARN mensajero tuvo un rendimiento de 0,53µg/mg de tejido cardiaco y una relación DO260/DO280 de 1,8. En la electroforesis en gel de agarosa con formaldehído, aparecieron las bandas de gran pureza correspondientes al ARN. Este protocolo permitió contar con material suficiente para obtener ADNc y aumentar el rendimiento tres veces con respecto al obtenido por Chomczinski y cols. [10]. En la reacción de la transcriptasa inversa y la RCP con el ARN total del tejido cardiaco, se obtuvo una banda de 631 pares de bases (pb) que coincide con la reportada para el ADNc de la TnIc (Figura 1). Figura 1. Electroforesis en agarosa de ADN complementario obtenido a partir de ARN de tejido cardiaco a través de la reacción de la transcriptasa inversa seguida de la RCP. Carrilera 1, control negativo; carrilera 2, gen amplificado (631 pb); PM, marcador de peso molecular (1 Kb DNA ladder, GIBCO-BRL, EUA). Mediante hibridación de colonias se detectaron 14 clones positivos, de los cuales 7 expresaron la TnIcr a altos niveles. De los 7 clones, 2 tuvieron niveles de expresión de hasta 43% de las proteínas celulares totales: pRTI4.5 y pRTI2.8 (Figura2A). Estos dos clones fueron secuenciados parcialmente en la región final del gen de la proteína de fusión, con lo que se confirmó la inserción en el marco de lectura y la presencia de dos mutaciones puntuales del gen de la TnIc [18]. Las dos mutaciones encontradas coinciden con las reportadas previamente por Armour y cols. [8] y Hayden y cols. [9], al comparar sus secuencias con la de Vallins y cols. [15]. Una de las mutaciones consiste en una simple transversión de G por C que resulta en un cambio silente en la posición Arg192 de la TnIc, y la otra es una transición de G por A que provoca un cambio aminoacídico de Ala por Thr en la posición 86. Estas mutaciones sugieren la existencia de dos isoformas de la TnIc humana. Se obtuvieron aproximadamente 3,3g de biomasa húmeda de E.coli por litro de cultivo inducido de la TnIcr, y 2,9g de biomasa húmeda de E.coli por litro de cultivo inducido del fTnIcr. La TnIcr y el fTnIcr se expresaron a altos niveles como proteínas insolubles. Los niveles de expresión del fragmento recombinante correspondieron a 30% de las proteínas celulares totales (Figura2B). La secuencia del fragmento recombinante coincidió con las reportadas anteriormente para esa región [8,9,18]. Figura 2. A) Electroforesis en gel de poliacrilamida 12% de los clones que expresan la TnIcr. Carrilera 1, pRTI2.8; carrilera 2, pRTI4.5; carrilera 3, control negativo; carrilera 4, TnIc natural. B) Electroforesis en gel de poliacrilamida 15% del clon que expresa el fTnlcr. Carrilera 1, pRfTI4.5; carrilera 2, control negativo; PM, marcador de peso molecular. Purificación de las proteínas recombinantes Las dos proteínas recombinantes se trataron de solubilizar en urea 2M, 4M, 6M, 8M y 9M, así como en cloruro de guanidio a esas mismas concentraciones. Se obtuvo una solubilización parcial a partir de 6M (datos no mostrados). Esto permitió la realización de lavados de urea 4M y la obtención de un buen grado de pureza para las dos moléculas recombinantes. Con el propósito de aumentar la pureza y de obtener una proteína soluble, se realizó la electroelución y se obtuvieron 12,4mg de TnIcr (peso húmedo) y 7,8mg de fTnclr por gramo de biomasa de E.coli. El grado de pureza obtenido para la proteína entera y su fragmento fue de 94% y 92%, respectivamente. En la Figura 3, se muestra el resultado de SDS-PAGE de la TnIcr y el fTnIcr en condiciones reductoras (b-mercaptoetanol 0,1%) y en condiciones no reductoras (sin b-mercaptoetanol 0.1%), lo que evidencia el elevado grado de pureza de ambas moléculas. Las bandas por debajo de 30kD (Figura 3A), sugieren alguna degradación proteolítica de la TnIcr. También se puede observar que el peso molecular de la TnIcr es superior al de la TnIc natural, debido a la presencia de la secuencia estabilizadora de IL-258 en el extremo amino y las seis histidinas del extremo carboxilo de la proteína recombinante. En ambas electroforesis en presencia de condiciones no reductoras, se observa una banda tenue para la Tnlcr y el fTnlcr, correspondiente a proteínas con pesos moleculares aproximados de 68kD y 29 kD, respectivamente, que pudieran ser dímeros de las moléculas recombinantes. Figura 3. Electroforesis desnaturalizante en gel de poliacrilamida 12% para la purificación de la TnIcr por electroelución (A), y en gel de poliacrilamida 15% para la purificación de fTnIcr (B). Carrilera 1, condiciones no reductoras; carrilera 2, sedimento de ruptura celular lavado con urea 4M; carrilera 3, condiciones reductoras; PM, patrón de peso molecular. Inmunoreactividad de las proteínas recombinantes La mezcla de sueros de ratones inmunizados con la TnIcr, reconoció a la TnIc natural y a la Tnlc recombinante en un ensayo de ultramicroELISA (Figura 4A). Este resultado indica que la TnIcr expone epitopos similares a los de la TnIc natural. La mezcla de sueros de ratones inmunizados con el fTnIcr reconoció a la TnIc natural y, en menor medida, al fragmento recombinante (Figura 4B). Una posible explicación puede ser que el fragmento aminoterminal está mejor expuesto en la TnIc natural en su unión con la fase sólida, comparado con el fTnIcr, debido al mayor peso molecular de la proteína natural con respecto al fragmento recombinante. También se encontró una respuesta de anticuerpos contra el fragmento recombinante de la IL-2 (Figura4), pero no con la intensidad encontrada para las dos proteínas recombinantes y la TnIc natural. Figure 4. ELISA de reactividad de los sueros de ratones inmunizados con TnIcr (A) y fTnIcr (B), contra la TnIc natural (TnIcn) y la IL-2 humana recombinante. Los valores utilizados en la realización de los gráficos son la media de dos determinaciones. Las barras representan +/- 2 desviaciones estándar. Discusión La selección de un sistema de expresión para la producción de una proteína recombinante en E. coli, depende del objetivo final que se persigue. Se prefiere la estrategia de utilizar proteínas de fusión en la producción de proteínas recombinantes, cuando se desea inducir una respuesta de anticuerpos contra una proteína en particular [19]. El objetivo de este trabajo fue la obtención de TnIc y su fragmento aminoterminal recombinante, para utilizarlos posteriormente como inmunógenos en la generación de anticuerpos monoclonales (AcM) cardioespecíficos contra la región aminoterminal o contra la molécula completa de la TnIc. Existen tres vías fundamentales para la obtención de la troponina con vistas a utilizarla como inmunógeno: la natural, la comercial y la recombinante. La primera consiste en la purificación de la TnIc humana a partir de su fuente natural, pero este procedimiento tiene problemas prácticos que dificultan el proceso: 1) la baja solubilidad de la TnIc natural en tampones de baja fuerza iónica; 2) la presencia de dos residuos de cisteína en su secuencia con extrema susceptibilidad a la oxidación; y 3) la extrema susceptibilidad de la TnI a la proteólisis durante su aislamiento de tejido cardiaco, con la formación de una proteína truncada de 14kD [9,20]. La TnIc natural se puede adquirir por la vía comercial, pero es un reactivo caro (0,1mg/450.00 USD). La tecnología del ADN recombinante es una vía alternativa que tiene valor porque permite disponer de un vector a partir del cual se han logrado altos niveles de expresión de proteínas recombinantes fusionadas al fragmento de IL-258, y porque se pudo obtener el fragmento aminoterminal cardioespecífico de la TnIc, con vistas a dirigir la respuesta de anticuerpos de ratón hacia esa zona de la troponina. Esta última vía pudiera tener la desventaja de que se genere en los ratones una respuesta inmune más intensa contra el fragmento de IL-258 que contra la TnIc que forma parte de las proteínas recombinantes de fusión. Como se discutirá más adelante, esto no sucedió. En dos trabajos anteriores, se ha obtenido TnIcr utilizando una proteína de fusión mucho mayor que la TnIc. En uno de ellos, no se informó el rendimiento obtenido [3] y, en el otro, se obtuvo una expresión de sólo 8% de las proteínas celulares totales [4]. En esos mismos trabajos, se utilizaron secuencias estabilizadoras que representan 84% de la proteína de fusión, como, por ejemplo, la b-galactosidasa, y 50% en el caso de la CMP-KDO sintetasa. En este trabajo, el fragmento de IL-258 representa 20% de la proteína de fusión con la TnIcr y 65% de la proteína de fusión con el fTnIcr, y en ambos casos se obtuvieron niveles de expresión mayores que los reportados. Lo anterior sugiere que el por ciento que representa el tamaño de la secuencia estabilizadora en la proteína de fusión, no repercute en los altos niveles de expresión de la TnIc ni de su fragmento aminoterminal. La expresión se realizó a partir del plasmidio pR2M6, que contiene el promotor triptófano. Este vector derivado del pFP-15 [10], fue diseñado para la producción de antígenos recombinantes de diagnóstico in vitro como proteínas de fusión con un dominio de 58 aa de la IL-2 humana en el extremo aminoterminal, y una secuencia codificante de 6 histidinas en el extremo carboxilo terminal. El fragmento de fusión de la IL-2 humana, aumenta la expresión de antígenos recombinantes por un mecanismo aún no dilucidado. Este plasmidio ha sido utilizado para la expresión de cerca de 10 proteínas recombinantes de los virus de inmunodeficiencia humana tipo 1 y 2, los virus 1 y 2 del linfoma/leucemia de células T, el virus de la hepatitis B y la proteína TmpA de Treponema pallidum. Los niveles de expresión han oscilado entre 10% y 30% de las proteínas bacterianas totales [21]. En este trabajo, se reafirma la consistencia en los altos niveles de expresión de este vector para la TnIcr y el fTnIcr, así como la estabilidad de la expresión de proteínas heterólogas que aporta la secuencia IL-258 en E.coli. Los rendimientos de 41mg de TnIcr y 23mg de fTnIcr purificados por litro de cultivo de E.coli, no son tan altos como los reportados por Davies y cols. [22] de 50mg de TnIcr/L, pero constituyen una fuente estable de TnIcr y fTnIcr que cubre ampliamente las necesidades inmediatas de investigación en los laboratorios donde se realizan estos trabajos. Resulta curioso que para ambas proteínas recombinantes, aunque presentan un nivel de expresión similar, los rendimientos finales son diferentes. Los rendimientos en el cultivo antes de la electroelución eran similares para ambas proteínas: 3,3g de biomasa húmeda de TnIcr/L de cultivo de E. coli y 2,9 g de biomasa húmeda de fTnIcr/L de cultivo de E.coli. Por lo tanto, la diferencia en el rendimiento final entre las dos proteínas recombinantes se manifiesta después de la electroelución. Posiblemente, el fragmento recombinante sea más susceptible a la degradación proteolítica por su menor talla, lo que origina fragmentos con pesos moleculares menores de 10kD que se pierden durante la diálisis. Con la electroelución, se obtuvo un alto grado de pureza para ambas moléculas. Sin embargo, en la SDS-PAGE de ambas proteínas recombinantes en condiciones no reductoras, aparecieron bandas de proteínas con una talla aproximadamente del doble, lo que puede corresponder a la formación de dímeros del fragmento y de la molécula completa. La TnIcr presenta tres residuos de cisteína: dos aportados por la TnIc natural [9] y otro codificado por el sitio XbaI introducido en la secuencia de ADN que codifica el fragmento de la IL-2 [10]. El fTnIcr presenta un residuo de cisteína codificado por el sitio XbaI, por lo que, en ambos casos, hay probabilidades de que se formen puentes disulfuro intermoleculares. La presencia de las cisteínas, la alta insolubilidad de estas proteínas recombinantes y sus altos niveles de expresión, sugieren la formación de cuerpos de inclusión después de la inducción con ácido b-indolacrílico. El por ciento de homología entre la Tnlc humana y la Tnlc de ratón es elevado. Por ejemplo, de los 210 aa que poseen, 195 son idénticos en ambas proteínas y 9 de los 15 aa restantes son conservados [23]. A pesar de la gran homología, tanto la TnIcr como el fTnIcr resultaron ser buenos inmunógenos, debido a que se obtuvieron títulos elevados de anticuerpos en los ratones inmunizados. El reconocimiento de la TnIc natural por el suero de un ratón inmunizado con TnIcr, sugiere que hay similitud en los epitopos secuenciales de ambas moléculas, puesto que no se puede hablar de epitopos conformacionales porque ambas troponinas se encuentran desnaturalizadas: la TnIcr en SDS y la TnIc natural en urea 8M (instrucciones del fabricante). También, en el caso del fTnIcr se obtuvo una buena respuesta antiTnIc natural. Es necesario resaltar que el fragmento de la IL-2 humana genera títulos de anticuerpos relativamente bajos con respecto a la secuencia de la TnIc de ambas proteínas recombinantes. Este hecho señala que la secuencia de 58 aa de la IL-2 humana no presenta epitopos inmunodominantes de interés. La antigenicidad de estas moléculas recombinantes es bastante cercana a la de la TnI natural, por lo que constituyen un buen inmunógeno para generar AcM contra la TnIc. Estos anticuerpos serían herramientas básicas para el desarrollo de estuches de reactivos de diagnóstico para la detección del infarto agudo de miocardio. Hasta el momento, y con el empleo de la TnIcr y el fTnIcr como inmunógenos, los autores de este trabajo han obtenido tres AcM antiTnIc cardiospecíficos y cuatro no cardioespecíficos que se encuentran en fase de caracterización y evaluación en la actualidad. También, la TnIcr puede emplearse en el control del funcionamiento de estos estuches de reactivos. Agradecimientos Agradecemos al Dr. Jorge Gavilondo la idea de la obtención de la troponina I por la vía recombinante, al Dr. Carlos Martínez por suministrar el corazón humano, y al Lic. Alexis Mussachio y al Dr. Juan Roca por participar en la discusión de algunos experimentos. Referencias 1. Cummins P, Young A, Auckland ML, Michie CA, Stone PCW, Shepstone BJ. Comparison of serum cardiac specific troponin I with creatine kinase, creatine kinase-MB isoenzyme, tropomyosin, myoglobin and C-reactive protein release in marathon runners: cardiac or skeletal muscle trauma. Eur J Clin Invest 1987;17: 317-24. 2. Haider KH H, Stimson WH. Production and characterisation of anti-cardiac troponin I monoclonal antibodies. Disease Markers. 1994;12:187-97. 3. Bhayana V, Henderson AR. Biochemical markers of myocardial damage. Clinical Biochemistry 1995;28:1-29. 4. Mair J, Wagner I, Morass B, Fridrich L, Lechleitner P, Dienstl F, et al. Cardiac troponin I release correlates with myocardial infarction size. Eur J Clin Chem Clin Biochem 1995;33:869-72. 5. Kragten JA, Hermens WTH, van Dieijen-Visser MP. Cardiac troponin T release into plasma after acute myocardial infarction: only fractional recovery compared with enzymes. Ann Clin Biochem 1996;33:314-23. 6. Willging S, Keller F, Steinbach G. Specificity of cardiac troponins I and T in renal disease. Clin Chem Lab Med 1998; 36:87-92. 7. Ebashi S, Wakabayashi T, Ebashi F. Troponin and its components. J Biochem 1971;69:441-5. 8. Armour KL, Harris WJ, Tempest PR. Cloning and expression in Escherichia coli of the cDNA encoding human cardiac troponin I. Gene 1993;131:287-92. 9. Hayden M, Traphagen L, Wilkins J, Schmitz E, Laird D, Herrmann R, et al. Expression in Escherichia coli and affinity purification of a CKS-troponin I fusion protein. Protein expression and purification 1995;6:256-64. 10. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thyocyanate-phenol-chloroform extraction. Anal Biochem 1987;162: 156-9. 11. Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: A laboratory manual New York: Cold Spring Harbor Laboratory Press; 1989. 12. Rodríguez-Tomé P, Stoehr PJ, Cameron GN, Flores TP. The European Bioinformatics Institute (EBI) databases. Nucl Acid Res 1996;24:6-13. 13. Novoa LI, Machado J, Fernández JR, Benitez J, Narciandi RE, Reinoso JL, et al. Heber Biotec, assignee. Method for the expression of heterologous proteins produced in fused form in E.coli, use thereof, expression vectors and recombinant strains. European Patent Application 90202108.8.,02.08.90. 14. Sanger F, Nicklen S, Coulsen AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci 1977;74:5463-7. 15. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227: 680-5. 16. Fernández-Patrón C, Castellanos-Serra L, Rodríguez P. Reverse stainning of sodium dodecyl sulfate polyacrylamide gels by imidazole-zinc salts: Sensitive detection of unmodified proteins. Biotechniques 1992;12:564. 17. Harlow E, Lane D. Antibodies: A laboratory manual Eds. Cold Spring Harbor Laboratory Press, NY. 1988: 673. 18. Vallins WJ, Brand NJ, Dabhade N, Butler-Browne G, Yacoub MH, Barton PJR. Molecular cloning of human cardiac troponin I using polymerase chain reaction. FEBS letter 1990;270:57-61. 19. Bernard A, Payton M. Selection of E.coli expression systems. In: Colligan JE, Dum BM, Ploegh HL, Speicher DW, Wingfield PT, editors. Current Protocols in Protein Sciences. Vol. 1. Seattle: John Whiley & Sons Inc, 1995;5.2.1-18. 20. Katrukha AG, Bereznikova AV, Esakova TV, Filatov VL, Bulargina TV, Gusev NB. A new method of human troponin I and troponin T purification. Biochem and Mol Biol In 1995;36:195-202. 21. Miranda A, Benítez J, Acevedo B, Domínguez MC, Villareal A, Palenzuela D, et al. Serodiagnosis and immunotherapy in infectious diseases 1997;8:149-55. 22. Davies E, Gawad Y, Takahashi M, Shi Q, Lam P, Styba G, et al. Analytical performance and clinical utility of a sensitive immunoassay for determination of human troponin I. Clin Biochem 1997; 30:479-90. 23. Smith SC, Ladenson JH, Mason JW, Jaffe AS. Elevations of cardiac troponin I associated with myocarditis. Experimental and clinical correlates. Circulation 1997;95:1-6. Copyright 2000 Elfos Scientiae The following images related to this document are available:Line drawing images[ba00005b.gif] [ba00005a.gif] [ba00005d.gif] [ba00005c.gif] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}