 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
ARTICULO DE REVISION / REVIEW ARTICLE METODOS ESPECTROMETRICOS DE MASAS PARA EL ESTUDIO DE PROTEINAS Vladimir Besada, Luis J. Gonzalez y Gabriel Padron. Centro de Ingenieria Genetica y Biotecnologia, Apartado 6162, La Habana 6, Cuba. Recibido en mayo de 1995. Aprobado en mayo de 1995.
Code Number: BA95050
Sizes of Files:
Text: 52K
Graphics: Line Drawings (gif) 77KKey words: Mass espectrometry, ionization, sequencing, proteins, biomolecules, post-translational modifications.
SUMMARY The development of new ionization methods has allowed the establishment of mass spectrometry as analytical tool for the characterization of biomolecules, becoming an essential technique for the detection of modified amino acids in natural and recombinant proteins. Besides, this technique constitute a promising alternative for peptide and protein sequencing. Here, we describe the development of this technique in the last 20 years and the most advanced equipments nowadays used for protein studies. RESUMEN El desarrollo de nuevos metodos de ionizacion ha permitido a la espectrometria de masas establecerse como tecnica de analisis de biomoleculas. Actualmente constituye una tecnica insustituible para la deteccion de aminoacidos modificados en proteinas naturales y recombinantes y una alternativa prometedora para la secuenciacion de peptidos y proteinas. En este trabajo se describe el desarrollo de esta tecnica en los ultimos 20 anos y la instrumentacion mas empleada actualmente para el estudio de proteinas.
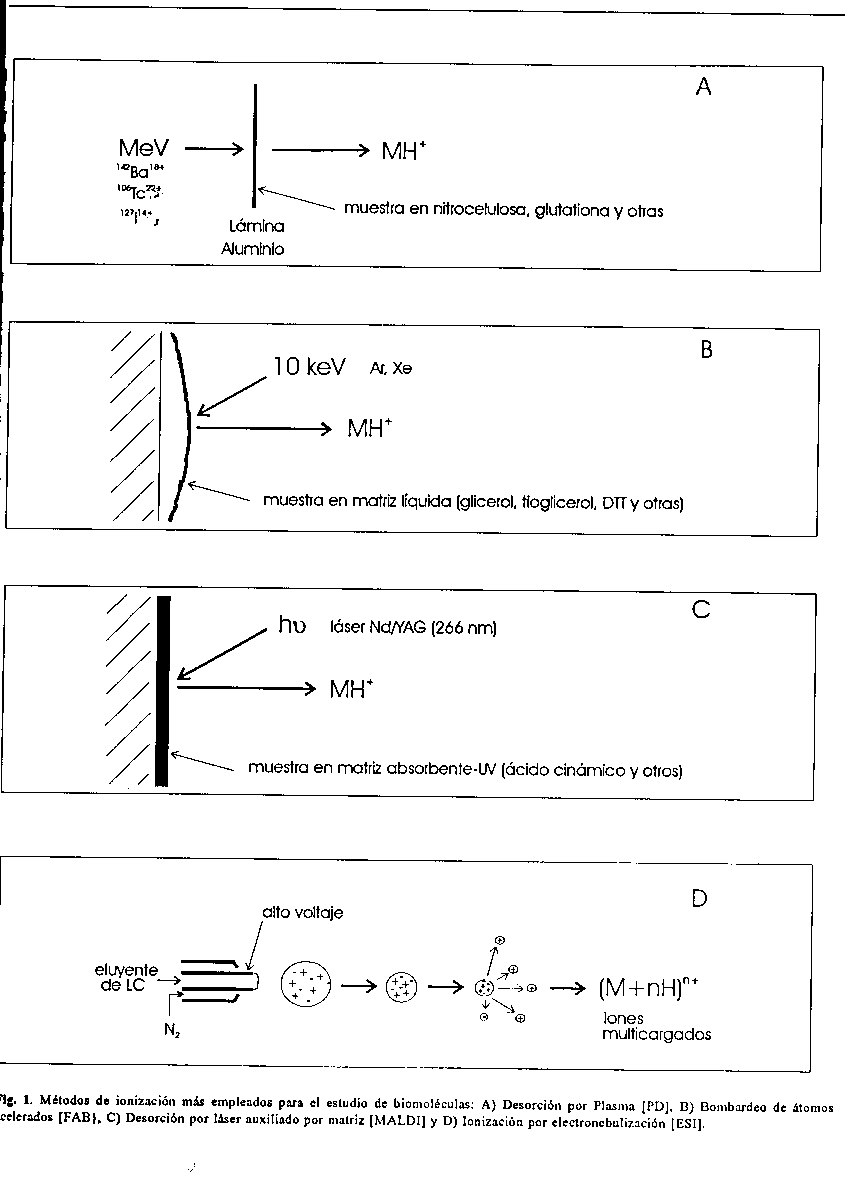
Desde la decada del 50, la espectrometria de masas (EM) era ya una reconocida herramienta en la elucidacion de estructuras de compuestos organicos(1), caracterizada por una alta exactitud en las mediciones y una alta sensibilidad. Sin embargo, hasta la decada del 80 la contribucion de esta tecnica a la elucidacion de la estructura primaria de las proteinas fue limitada(2, 3), pues aun para peptidos pequenos se requiere de una extensa derivacion (para conferirles volatilidad) y de sucesivos pasos de extraccion o purificacion del derivado. Estas razones, hicieron impracticable esta tecnica como via para la secuenciacion de proteinas de forma rutinaria. En los ultimos anos se han logrado dos avances importantes que permiten la aplicacion progresiva de la espectrometria de masas a las biomoleculas y en particular a las proteinas. En primer lugar, el diseno de novedosos metodos de ionizacion y en segundo lugar, la introduccion de mejoras tecnologicas en los analizadores ya existentes. METODOS DE IONIZACION La introduccion de nuevos metodos de ionizacion, que no requieren de la volatilizacion previa de la muestra, fue el elemento principal que permitio la extension de la EM al campo de las biomoleculas. Estos metodos provocan simultaneamente, tanto la ionizacion como la desorcion de las moleculas llevando a fase gaseosa iones pseudomoleculares del tipo (M+H)^+ y (M-H)^-. Sin embargo, la energia suministrada en estos casos genera una escasa fragmentacion del ion molecular, en donde se observa este mayoritariamente, razon por la que se le conoce como metodo "suave" de ionizacion. La tecnica de desorcion por campo electrico (FD)(4) fue la primera comercialmente disponible y consta de un alambre fino de tungsteno (1-5 m), previamente activado con carbono o silicio, por formacion de microagujas, sobre el cual se deposita la muestra en solucion y se somete a un alto voltaje (6-10 kV). El alto campo electrico y la temperatura generados provocan la ionizacion y desorcion de las moleculas. Este metodo si bien permitio determinar, por primera vez, la masa molecular de peptidos relativamente grandes sin derivacion alguna y secuenciar oligopeptidos posterior a tratamientos con exopeptidasas (5) y ciclos de degradacion de Edman (6), no demoro mucho en quedar aventajado por otros metodos, fundamentalmente debido a la trabajosa preparacion del filamento. No es hasta la introduccion de los metodos de ionizacion con haces de particulas que la EM se afianza como tecnica de analisis de biomoleculas. En 1974 MacFarlane y col. (7) emplearon fragmentos de alta energia (MeV), productos de la fision del ^252Cf, para bombardear aminoacidos libres. La fision del ^252Cf produce dos fragmentos con energias de 80 y 100 MeV (8). En este metodo, conocido por desorcion por plasma (PD), uno de los fragmentos llega a un detectowque marca el inicio de la medicion, y el otro, generado en sentido contrario, atraviesa la lamina metalica sobre la que esta depositada la muestra, elevando localmente la temperatura en el lugar del impacto, de forma tal que ocurre la desorcion e ionizacion del compuesto (figura 1A). Los fundamentos del proceso de ionizacion han sido abordados en diferentes trabajos sobre la tecnica (9, 10, 11).
Sin embargo, se ha reportado un fenomeno de supresion de las senales en mezclas de peptidos segun la carga neta de cada molecula (en dependencia de la composicion de aminoacidos basicos y acidos), o sea, los peptidos con carga neta positiva predominan en el espectro de iones positivos, cuando se analizan en presencia de peptidos con carga neta negativa y viceversa (14). Las principales limitantes del metodo estan en su sensibilidad, que no permite detectar cantidades de peptido inferiores a los cientos de picomoles (15) y en su bajo poder de resolucion, dado fundamentalmente por el tipo de analizador que emplea. De las tecnicas de desorcion de iones, la que mas ha sido empleada hasta el momento es la ionizacion por bombardeo con atomos rapidos (FAB), descrita en 1981 por Barber y col (16).Esta consiste en el bombardeo de la muestra con atomos de elevada energia cinetica (8-10 keV), que provoca la dispersion hacia la fase gaseosa de los iones moleculares preformados o de moleculas neutras que son posteriormente ionizadas (fig. 1B). El mecanismo de formacion de los iones ha sido discutido en diversos trabajos (17, 18). Segun algunos investigadores (19, 20) la ionizacion por bombardeo con un haz primario neutro no fue tan novedosa como la introduccion de matrices liquidas de baja tension de vapor como soporte de la muestra. La ventaja consiste en que la superficie donde incide el haz neutro es constantemente renovada, lo que posibilita obtener iones durante un tiempo prolongado, a diferencia de cuando se emplean matrices solidas, rapidamente deteriorables en pocos segundos. El glicerol es la matriz mas empleada para la mayoria de las muestras, debido a su baja presion de vapor, alta viscosidad, buena capacidad como solvente y bajo peso molecular (21). La hidrofobicidad de los peptidos influyen en el espectro obtenido (22), de modo que se ha considerado un grupo grande de liquidos como matriz o como aditivo del glicerol(23, 24), tales como la tioglicerina (25), una mezcla de ditiotreitol y ditioeritritol (26, 27). Sin embargo, el empleo de matrices liquidas trae aparejado un efecto de supresion de senales para mezcla de peptidos de diferente hidrofobicidad (28, 29, 30), lo cual impide generalmente, el empleo de esta tecnica para analisis cuantitativos. Desde la introduccion de este metodo se han reportado la caracterizacion y secuenciacion de numerosas proteinas naturales y recombinantes (31, 32). La EM se convirtio, por primera vez, en una tecnica imprescindible para la deteccion de modificaciones postraduccionales (33, 34). Esta tecnica tiene ventajas sobre las anteriores descritas, por la facil preparacion de la muestra y la capacidad de producir a temperatura ambiente un espectro de senales intensas y estables en el tiempo. Su principal limitante radica en su relativa baja sensibilidad, lo que hace que disminuya bruscamente, al incrementarse la masa. Si bien las tecnicas de ionizacion por PD y FAB surgieron en la misma etapa y la primera aventaja al FAB en alcance de masas (50 000 Da vs. 20 000 Da), solo esta ultima logro imponerse como metodo rutinario en muchos laboratorios del mundo, dado entre otros factores por su facil introduccion en los espectrometros ya existentes. No obstante, en los ultimos 5 anos los esfuerzos se han focalizado sobre disenos que permitan incrementos de sensibilidad y de alcance de masas, lo que ha impulsado el desarrollo de nuevos metodos de ionizacion (tabla 1). Tabla 1
-----------------------------------------------------------------
Metodo Valor de Ventajas
de m/z (Da)
ionizacion alcanzado
-------------------------------------------------------------
----
PD < 50 000 Sencillo de operar; permite analisis de
mezclas FAB < 20 000 Permite analisis de mezclas; da
informacion de secuencia y masa molecular de
peptidos; adaptable para CL/EM
MALDI< 450 000 Los mayores valores de masas alcanzados;
permite analisis de muestras; es relativamente
insensible a sales; de simple operacion;
util en glicoproteinas; sensibilidad femtomolar
ESI < 200 000 Permite analisis de mezclas; da informacion
de secuencia para peptidos pequenos puros;
facilmente adaptable a CL/EM y equipos de
cuadripolos; sensibilidad femtomolar,
exactitud < 0,01%
----------------------------------------------------------------
Metodo Valor de Desventajas
de m/z (Da)
ionizacion alcanzado
-----------------------------------------------------------------
PD < 50 000 Limites de resolucion muy bajos;
limitaciones para detectar variantes
estructurales; largos tiempos de
adquisicion > 1 h; no adaptado aun a
CL/EM; sensible a sales; sensibilidad
picomolar
FAB < 20 000 Interferencia de la matriz a m/z <
500; respuesta variable; efectos de
supresion inducida por peptidos; sensible
a sales; sensibilidad nanomolar
MALDI < 450 000 Limites de resolucion bajos; limitaciones
para detectar variantes estructurales;
informacion secuencial limitada; CL/EM aun
no establecida
ESI < 200 000 La carga multiple puede complicar la
interpretacion de los datos para mezclas;
no es util para glicoproteinas; sensible a
sales
-----------------------------------------------------------------
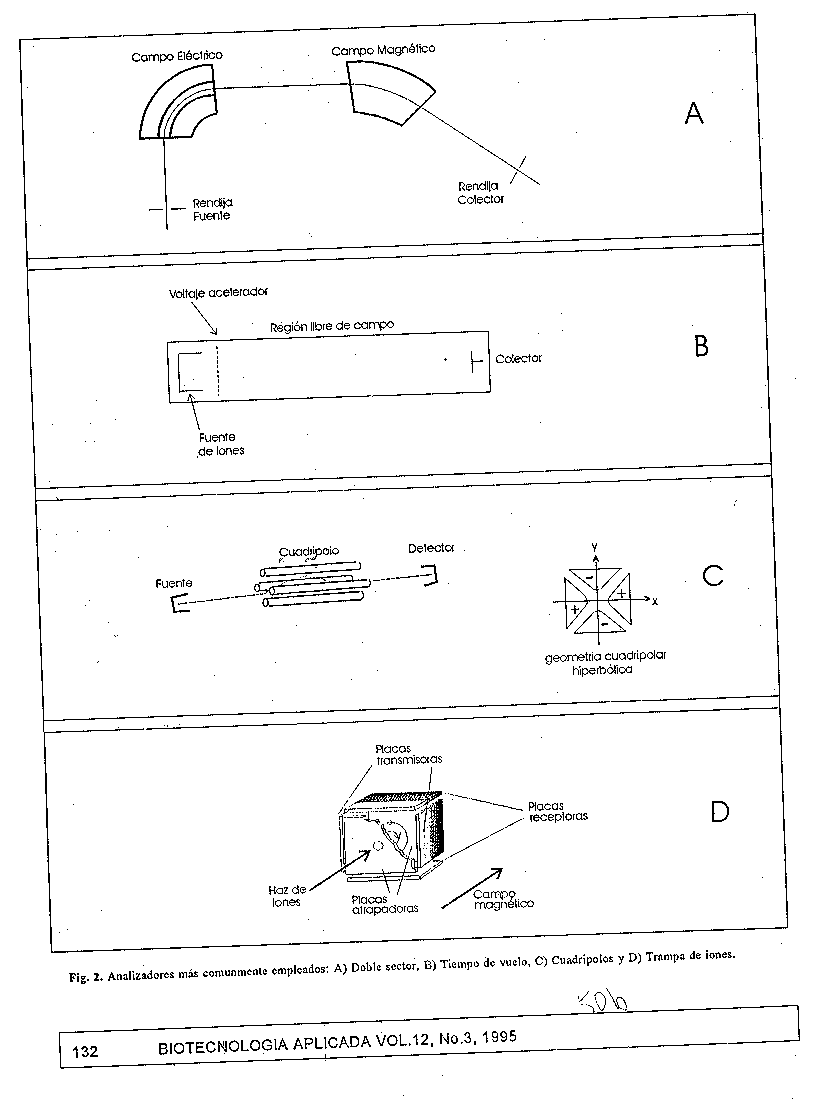
m/z- relacion masa-carga del ion CL- cromatografia liquida Entre estos metodos se encuentra la desorcion por laser, empleada por primera vez por Posthumus y col. (35) para el analisis de moleculas organicas no volatiles. Posteriormente se llego a obtener espectros de masas de moleculas grandes relativamente neutras, como oligosacaridos y glicolipidos, usando laser de CO2 y de Nd/YAG (36, 37). El laser, particularmente por pulso, es una fuente energetica atractiva para sustancias poco volatiles e inestables termicamente, pues se conoce que el calentamiento rapido de la muestra lleva con preferencia, a la evaporacion y no a la degradacion termica (38). No fue hasta finales de la decada del 80 que Karas y Hillenkamp, usando laser pulsado de UV y una matriz solida con fuerte absorcion en esta region del espectro (39, 40), lograron medir con alta sensibilidad proteinas (desde monomeros hasta tetrameros) con masas moleculares mayores de 100 000 Da (41). Entre estas estan la catalasa (monomerica de 236 000 Da), la ureasa (trimero de 275 000 Da) y un anticuerpo monoclonal (trimero de 450 000 Da) (39-42). El mecanismo mediante el cual ocurre la desorcion de la muestra se basa en la alta energia absorbida por la matriz, provocandose la perturbacion del sistema y transformandose en energia de excitacion asi como la desorcion de las moleculas de la muestra (38) (fig. 1C). La desorcion directa de las biomoleculas, sin el empleo de matriz, limitaba el analisis a masas moleculares menores de 1000 Da. El empleo de la matriz absorbente de la radiacion laser, da la posibilidad de analizar cualquier tipo de biomolecula independiente de su absorcion en el UV; y por otro lado, segun la energia depositada, permite obtener un espectro relativamente rico en fragmentaciones de la molecula (43). Numerosos compuestos organicos han sido propuestos para la preparacion de la matriz, entre ellos los mas empleados son: acido 2,5-dihidroxibenzoico (44, 15), alcohol 3- nitrobencilico (45, 46), acido cinamico y derivados de este (47, 48). Esta tecnica, conocida como metodo de desorcion e ionizacion por laser, auxiliado por matriz (MALDI), se caracteriza por una alta eficiencia de ionizacion y alta sensibilidad (en el orden de femtomoles). Algunas variaciones en el procedimiento de preparacion de la muestra ha permitido incrementos en sensibilidad (hasta el orden de attomoles) y en resolucion (49). Este metodo se ha empleado con exito en distintos tipos de moleculas no derivadas, entre las que se destacan, los oligosacaridos, las glicoproteinas (50) y los acidos nucleicos (51), estructuras estas dificiles de estudiar empleando cualquier otro metodo de ionizacion. MALDI, ademas, tiene la posibilidad de medir directamente proteinas electrotransferidas a membranas de PVDF, aun cuando estas hayan sido previamente tenidas (50, 53) o posterior a la proteolisis in situ (54). Otra de las tecnicas de desorcion de gran interes en la actualidad es la ionizacion por electronebulizacion (ESI), desarrollada por Fenn y cols (55) en 1989. En esta, la muestra en solucion entra a traves de un capilar a la camara de ionizacion a presion atmosferica, donde se establece una diferencia de potencial del orden de los kV entre la punta del capilar y el orificio de entrada al espectrometro. La combinacion del alto campo electrico en la punta y el flujo de nitrogeno gaseoso en dos sentidos, uno coaxial y el otro perpendicular al capilar, dispersa la corriente liquida en gotas muy pequenas que contenienen moleculas multicargadas (fig. 1D). La carga de los iones generados suele ser de tal magnitud que los valores de m/z a medir resultan muy bajos (< 2 500 Da), lo que ha permitido el empleo de los espectrometros de cuadripolos, de bajo alcance de masas. Esto posibilita la medicion de masas mayores de 100 000 Da como ya se ha demostrado en el estudio de la lactato deshidrogenasa (143 000 Da), de algunos anticuerpos monoclonales (150 000 Da) y del componente C4 del complemento humano (197 000 Da) (56, 57). Este metodo de ionizacion es de alta sensibilidad y la multiplicidad de las senales en el espectro reduce sustancialmente el error estadistico en la determinacion del valor de masa, lo que hace factible mediciones de muy buena exactitud (<0.01%). Recientemente, se logro con ESI detectar impurezas (mezcla de componentes proteicos minoritarios) por debajo del 3% en proteinas recombinantes (58), como los productos de degradacion de los extremos terminales N- y C- y de oxidacion de metionina, entre otras. Una de las grandes preocupaciones durante el desarrollo de estos metodos para el estudio de biomoleculas, ha sido encontrar la via adecuada para la introduccion de la muestra al espectrometro. Hasta el momento, la variante mas trabajada contempla el desarrollo de interfases entre la cromatografia liquida (HPLC) y el espectrometro, empleando fundamentalmente FAB (59, 60, 61, 62, 63) y en menor grado MALDI (46, 64)^.como metodo de ionizacion. Sin embargo, una de las principales ventajas del ESI es que, por diseno, es ya una interfase, lo que permite la caracterizacion directa de mezclas muy complejas, como es la de 200 peptidos enlazados a moleculas de clase I del complejo mayor de histocompatibilidad (MHC-I) (65). Este metodo de ionizacion ha resultado tambien, de gran interes para los estudios de estructura cuaternaria de las proteinas, pues la energia suministrada al sistema no parece afectar algunas interacciones no covalentes entre moleculas. Todas estas caracteristicas posibilitan su aplicacion en la determinacion de cambios conformacionales en las proteinas (66, 67, 68, 69), intermediarios en reacciones enzimaticas (70, 71, 72, 73), mecanismos de inhibicion (74), formacion de complejos enzimaticos covalentes (75) y no covalentes (76, 77, 78), entre otros muchos. Tanto MALDI como ESI permiten la determinacion facil de la integridad de las proteinas recombinantes y la obtencion de su mapa peptidico, asi como su rapida insercion entre las tecnicas empleadas para monitorear los procesos biotecnologicos y los productos asi obtenidos (79,80). Hasta hace diez anos el maximo valor de masa detectado no sobrepasaba los 15 000 Da, sin embargo la introduccion de estos dos ultimos metodos de ionizacion supero en mas de 30 veces ese valor. ANALIZADORES Actualmente los espectrometros que mas se comercializan tienen analizadores muy semejantes a los de hace 20 anos, con cambios menores en su arquitectura. Estos son los de doble enfoque, los de cuadripolo y los de tiempo de vuelo (TOF) (81) (tabla 2).
Tabla 2
-----------------------------------------------------------------
Doble sector Triple TOF
(tandem) Cuadripolo (reflectron)
-----------------------------------------------------------------
Alcance de masas Depende de V y B, <4 000 Da
Infinito(teorico)
125000 (teorica)
Sensibilidad Relativamente baja alta alta
Resolucion alta media/alta baja
Acoplable a: FAB,ESI,MALDI,CL ESI,CL PD, MALDI
Secuenciacion Generalmente permite
diferenciar aminoacidos
isobaricos No distingue
aminoacidos
isobaricos No totalmente
establecido
Costo aprox. >$1 500 000 $350 000 $300 000
-----------------------------------------------------------------
V- voltaje aceleradorB- intensidad de campo magnetico CL- cromatografia liquida Los analizadores de doble enfoque emplean dos sectores, uno electrico y otro magnetico (fig. 2A). El campo magnetico (B) es el encargado de enfocar el haz de iones monoenergetico (proveniente del campo electrico) hacia el detector segun la relacion masa/carga: m/z=B^2R^2/2V, donde R es el radio de curvatura del iman y V es el voltaje acelerador. Por tanto, el alcance de masas de estos equipos depende de la intensidad maxima del iman. Los nuevos espectrometros con analizadores de doble enfoque disponen de electroimanes muy potentes (hasta 2.3 Tesla), lo cual representa un alcance de hasta 125 000 Da al menor voltaje de aceleracion posible. Las mayores ventajas de estos analizadores estan dadas por su alta resolucion, su alcance de masas relativamente alto y la exactitud de las mediciones. Sus principales limitantes son la baja sensibilidad, el elevado costo y sus dimensiones.
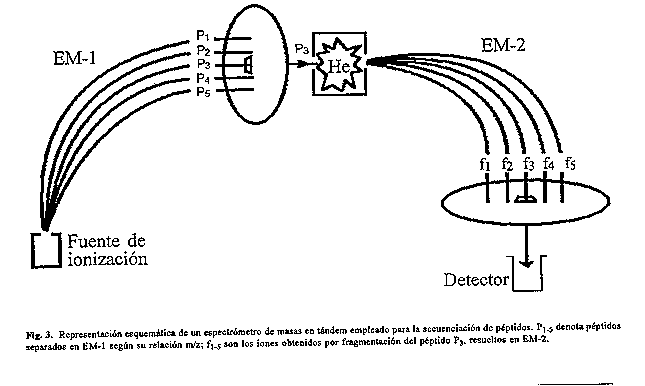
Por su lado, los analizadores de cuadripolo emplean cuatro electrodos, generalmente de seccion transversal hiperbolica (fig. 2B), entre los que se establecen voltajes de corriente directa y de radiofrecuencias (81). Para determinados valores de campo y para energias de traslacion menores de 100 eV, los cuadripolos funcionan como filtros de masas de alta selectividad. Estos analizadores son mas populares que los de doble enfoque debido a varias razones: son muy compactos, tienen alta velocidad de barrido y de sensibilidad. Sin embargo, comparados con los de doble enfoque, son de menor resolucion, alcance de masas y exactitud. Actualmente, la desventaja relacionada con el alcance de masas ha sido superada, pues la ionizacion por ESI permite medir directamente masas elevadas de moleculas multicargadas. La introduccion del metodo de ESI posibilito la incorporacion de los analizadores de cuadripolo al Arsenal de instrumentos, utilizados para el estudio de las proteinas. Los analizadores de tiempo de vuelo son los de arquitectura mas sencilla, pues emplean un simple tubo de aproximadamente un metro (fig. 2C), donde los iones son resueltos por el tiempo que demoran en recorrerlo, lo cual esta directamente relacionado con la masa de cada ion, segun: t=a(m/z)^1/2, donde a es una constante. Estos analizadores tienen un alcance de masas ilimitado y se caracterizan por su alta sensibilidad y velocidad de barrido. En los ultimos anos, la resolucion de estos analizadores ha mejorado notablemente debido a la introduccion de anillas reflectoras y un alargamiento en la trayectoria optica de los iones. La combinacion de los distintos metodos de ionizacion con los analizadores anteriormente descritos, ha generado diversidad de espectrometros entre los que se destacan: a) ESI o FAB acoplado al doble sector; b) ESI acoplado al cuadripolo; c) MALDI acoplado al analizador TOF, con diversas posibilidades de aplicacion. En dependencia del problema a investigar se selecciona el analizador y el metodo de ionizacion, teniendo en cuenta el rango de masas, el poder de resolucion, la exactitud en la medicion, la sensibilidad y la compatibilidad con otras tecnicas (como HPLC), entre otros aspectos. En la actualidad, se ha revitalizado el empleo de trampas ionicas como analizadores (fig. 2D), pues tiene grandes pespectivas de uso para el trabajo con biomoleculas (82). Existen varias caracteristicas que hacen superiores estos analizadores a los anteriormente descritos, como son: diseno mecanico muy sencillo (83), alta resolucion (84, 85), alta sensibilidad, compatibilidad con los distintos metodos de ionizacion^., capacidad para almacenar iones durante un largo periodo de tiempo (de mseg hasta minutos) y la deteccion simultanea de todos los iones presentes, seguido de una transformacion de Fourier (FT- MS) de los datos. Sin embargo a pesar de todas las ventajas mencionadas, aun existen problemas tecnologicos en el diseno de estos equipos que han impedido su comercializacion. Entre estos problemas se encuentran los relacionados con la manipulacion de iones de alta masa molecular y con el acoplamiento del ESI como metodo de ionizacion. ESPECTROMETRIA DE MASAS EN TANDEM (MS/MS) En el caso de las biomoleculas, uno de los grandes retos esta en la secuenciacion de peptidos y proteinas. Entre las configuraciones de equipos mas usadas con este fin estan: la disposicion en tandem de dos espectrometros de doble sector, el sistema cuadripolar triple y las trampas de iones (86,87,88) (tabla 2). En general, estos sistemas se disenan de tal forma que, en una primera etapa se selecciona el ion de interes (EM-1, fig. 3); en una segunda etapa, este ion se hace chocar con gas He o Ar para inducir su fragmentacion y los fragmentos de iones asi obtenidos se resuelven en una tercera etapa (EM-2).
En el caso de los espectrometros de doble sector, un ion precursor P3^+ (fig. 3) resuelto en EM-1 y los iones hijos que de este se deriven (f1^+, f2^+, f3^+, etc.), al ser generados en una region libre de campo, tienen velocidades muy semejantes con diferente energia cinetica. Por ello para lograr que tanto el precursor como sus iones hijos atraviesen el campo electrico y sean enfocados por el campo magnetico, atendiendo a su relacion m/z, debe realizarse un barrido simultaneo de los campos magnetico (B) y electrico (E) (linked-scan) (89), manteniendo constante la relacion B/E. El espectro asi obtenido da informacion solamente de los fragmentos de iones que se forman a partir de un unico ion precursor. Para detectar los fragmentos (hijos) provenientes de otro ion precursor, simplemente se selecciona otra relacion B/E, de acuerdo a su masa molecular. Esta tecnica permite la secuenciacion completa de peptidos de hasta 2 500 Da, de forma bidireccional (desde los extremos terminales N y C simultaneamente), incluso en mezclas de peptidos. La cantidad de peptido necesaria para obtener la completa informacion de la secuencia va desde 100 fmol hasta 1 nmol, dependiendo de la propia secuencia del peptido, del metodo de ionizacion y del analizador que se use. La ruptura de un simple enlace a lo largo de la cadena polipeptidica produce un fragmento cuya masa se corresponde con la suma de todas las cadenas laterales, mas el numero de unidades aminoacidicas contenidas en este (90). Por tanto, la interpretacion del espectro de masas de un fragmento peptidico (de una molecula desconocida) puede ser relativamente sencilla, teniendo en cuenta que las proteinas naturales estan compuestas por solo 20 aminoacidos. En la figura 4 se muestran las distintas series de iones que se forman por fragmentacion en el entorno de cada enlace peptidico (91). La diferencia de masas de series consecutivas de un mismo tipo nos brinda informacion de las masas de los aminoacidos dispuestos en un orden secuencial (92). Ademas, a diferencia de la secuenciacion automatica por degradacion quimica, permite detectar e identificar modificaciones quimicas en peptidos.
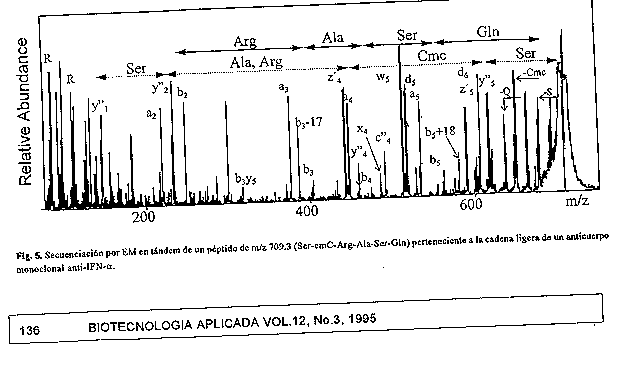
La aparicion e intensidad relativa de alguna de las series ionicas con respecto a otras depende, en gran medida, de la propia secuencia de aminoacidos (93), de la energia del haz de xenon (94), del gas de colision (95) y de los componentes de la matriz, entre otros factores. La presencia de aminoacidos basicos, particularmente arginina, en los extremos terminales N o C del peptido incrementan la intensidad de las series b, a, d o y", w, v, z, respectivamente, fenomeno conocido como charge- remote fragmentation (93, 96). Las senales correspondientes a la serie y" por corte en el extremo terminal N de Pro y a la serie b por perdida de Pro, tambien se intensifican (97). Por otro lado, es frecuente encontrar fragmentaciones de las cadenas laterales (series w, v, d y m) cuando se usa gas argon en lugar de helio (94). Igualmente se ha observado que, la adicion de acidos fosforico o sulfurico a la matriz, por ejemplo, produce mejor fragmentacion del peptido que cuando se usa HCl (98) En la figura 5 se muestra la secuenciacion de un peptido a partir del espectro obtenido en un equipo de doble sector en tandem con FAB acoplado.
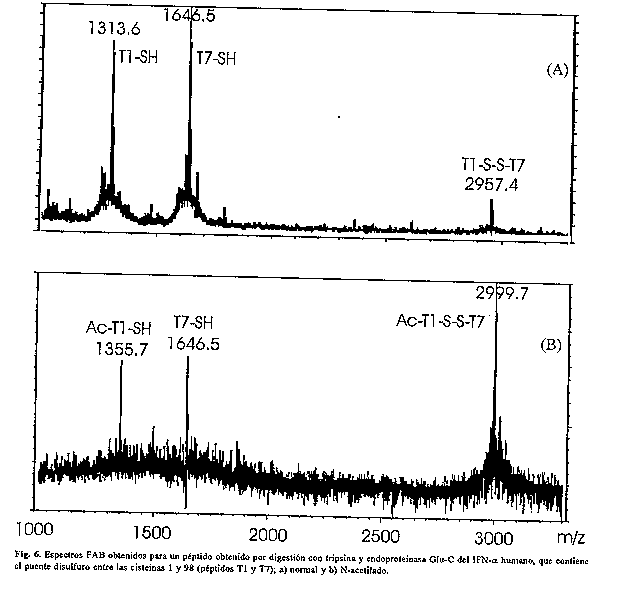
En la actualidad, sin embargo, los sistemas de triple cuadripolo se han impuesto sobre los de sector magnetico, por ser mucho mas baratos, mas sensibles, menos espaciosos y mas sencillos de operar. Por otro lado, los espectros obtenidos con el triple cuadripolo son mas sencillos de interpretar, pues al trabajar con iones de baja energia cinetica produce menos fragmentaciones que los de doble sector. No obstante, los sistemas de cuadripolo tienen la desventaja de no permitir la diferenciacion entre aminoacidos de igual masa como: leucina/isoleucina. Por otro lado, el empleo de FT-MS (con trampas ionicas) permite excitar, con pulsos apropiados de radiofrecuencias, un ion o grupo de iones determinado, realizar la fragmentacion del peptido de interes al provocar su colision con He o Ar y registrar los fragmentos obtenidos (99). De esta forma, repitiendo esta secuencia de pasos n veces es posible realizar experimentos de Ms^n (100), de gran utilidad en la elucidacion estructural de compuestos. Recientemente se ha reportado el empleo de anillas reflectoras en los equipos de MALDI-TOF para eliminar la diferencia energetica entre iones de igual masa. Aunque han habido escasas aplicaciones (43) del metodo a la secuencia de peptidos, este promete ser una tecnica de amplio uso. Como se observa, es posible aplicar la EM con disimiles propositos en el caso de proteinas, sin embargo actualmente la principal dificultad consiste en que no se dispone de un unico equipo que permita realizar todas las posibilidades de medicion; sino que, segun el problema concreto a resolver, se recurre a uno u otro espectrometro. APLICACIONES Las caracteristicas de los distintos metodos espectrometricos de masas permiten numerosas aplicaciones de la tecnica en el caso particular de las proteinas, sean de secuencias desconocidas o recombinantes. Existe una estrategia bien establecida para la caracterizacion de proteinas (101). Entre los ejemplos de aplicaciones mas referidas en la literatura estan: LOCALIZACION DE ENLACES DISULFURO Los enlaces disulfuro pueden localizarse empleando diferentes estrategias, sin embargo, el uso de la EM (a partir de la introduccion de FAB) facilita la manipulacion e identificacion de los peptidos enlazados por puentes S-S. El primer articulo que destaca la utilidad del FAB para este proposito fue publicado en 1985 (102); desde entonces se ha realizado la localizacion de enlaces disulfuro en numerosas proteinas. Los peptidos que contienen enlaces disulfuro se detectan en el espectro de masas por las senales de m/z correspondiente a ambos peptidos unidos y las de cada peptido reducido. En la figura 6 se ejemplifica con el espectro obtenido para un peptido de IFN- alpha que contiene el puente disulfuro entre las cisteinas 1 y 98. Se observan las senales del peptido original (m/z 2957.4) y la de ambos peptidos reducidos (m/z 1313.6 y 1646.5).
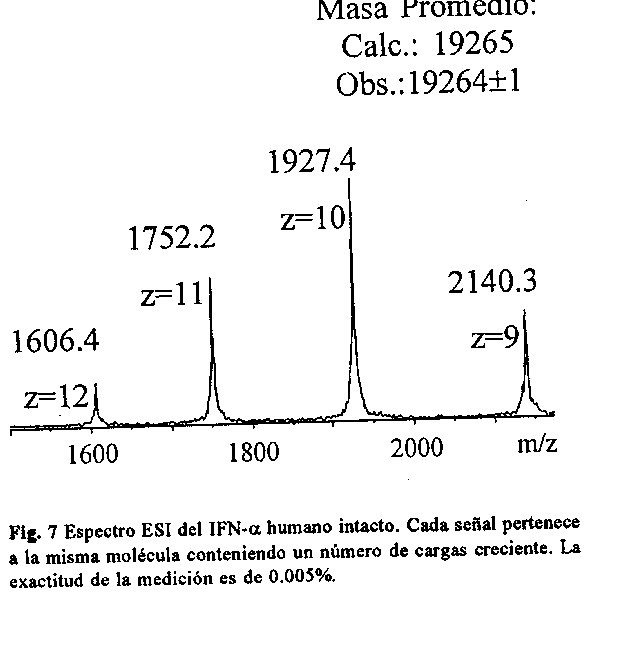
DETECCION DE MODIFICACIONES POSTRADUCCIONALES Posiblemente esta sea la aplicacion mas importante de la EM en el campo de las proteinas. El conocimiento de la secuencia de ADN que codifica para una proteina no es criterio suficiente para suponer la sintesis correcta, pues se conocen gran numero de ellas que se modifican durante o posterior a la traduccion del ARN mensajero. Se han reportado cerca de 200 modificaciones postraduccionales (34), por lo que la caracterizacion detallada tanto de las moleculas naturales como de las correspondientes proteinas recombinantes es absolutamente imprescindible cuando se trata de la produccion de farmacos para uso terapeutico (103, 104). En estos casos, la EM juega un papel esencial pues define directamente la estructura quimica de la modificacion. Una de las modificaciones mas frecuentes es la acetilacion del extremo terminal N de las proteinas, lo cual impide su secuenciacion. En la figura 6B se muestra la deteccion del IFN- alpha acetilado en el grupo amino terminal, por desplazamiento del valor de masa en 42 Da con respecto al IFN normal (fig. 6A). SECUENCIACION DE PROTEINAS Si bien existe una metodologia para la secuenciacion de las proteinas por via quimica desde su extremo terminal N, es tambien conocido que esta no es viable en moleculas con el grupo amino bloqueado (105) La secuenciacion por EM en tandem muestra algunos rasgos que aventajan a la via quimica, estos son: a) permite la secuenciacion bidireccional de los peptidos; b) permite la deteccion de aminoacidos modificados; c) la velocidad de secuenciacion es mucho mayor; d) mayor sensibilidad; e) no requiere de una purificacion exhaustiva de la muestra, por lo que permite trabajar con mezclas de peptidos (90). Existen numerosos ejemplos de proteinas secuenciadas por espectrometria de masas. No obstante, la EM no sustituye a la tecnica de secuenciacion automatica por via quimica, sino que la complementa; de modo que el uso combinado de ambas facilita la caracterizacion estructural de la proteina (106). MAPEO PEPTIDICO Este se realiza normalmente por HPLC en columnas de fase inversa y constituye uno de los ensayos que se exige a los productos farmaceuticos para definir su identidad (107). Actualmente, la introduccion de los metodos de ionizacion por ESI y MALDI, permiten con relativa facilidad, un analisis mas completo de la muestra, al generar no solo el mapa peptidico sino tambien, la masa molecular de cada peptido. DETERMINACION DEL EXTREMO TERMINAL C Aun no existe una metodologia establecida que permita la secuenciacion de las proteinas por su extremo terminal C, aunque si se han reportado varios procedimientos para la deteccion del ultimo aminoacido o del peptido correspondiente al extremo C. En el primer caso, se detecta el aminoacido libre posterior a hidrazinolisis (108) o tritiacion (109) de la proteina. La via enzimatica empleando diversas carboxipeptidasas tambien ha sido reportada (110), sin embargo funciona preferiblemente en peptidos y resulta poco util para proteinas. Mientras en el segundo caso, se detecta el peptido liberado del extremo C, por afinidad de las enzimas anhidrotripsina y anhidroquimotripsina (111) a los peptidos con lisina. Otra via que se emplea, considera la funcion de transpeptidacion de la carboxipeptidasa marcan el peptido correspondiente, luego de eliminar el ultimo aminoacido (112). Otra alternativa lo constituye el empleo de agua enriquecida con ^18O durante la digestion enzimatica de la proteina. Esto permite la deteccion por EM del peptido correspondiente por aparicion de una senal con distribucion isotopica normal (113, 114), a diferencia de la observada para los peptidos marcados. IDENTIFICACION DE PROTEINAS La identificacion de proteinas en extractos naturales es extremadamente laboriosa y compleja. La EM juega un papel muy importante en el caso de proteinas bloqueadas en el extremo terminal N. Existen diferentes procedimientos para lograr la identificacion, que incluyen: 1) mapeo peptidico con enzimas especificas y busqueda en el banco de secuencias por comparacion de los valores de masas observados (115, 116). 2) busqueda en el banco de secuencias a partir de las fragmentaciones observadas en el espectro MS/MS (117). 3) secuenciacion de un peptido interno por MS/MS, determinacion de la masa molecular de la proteina intacta y busqueda de semejanza en el banco de secuencias. DETERMINACION DE LA MASA MOLECULAR DE PROTEINAS INTACTAS CON GRAN EXACTITUD La tecnica de electroforesis (SDS-PAGE) continua siendo la mas empleada para la determinacion de la talla molecular de las proteinas, por su facil instrumentacion en cualquier laboratorio. Con esta se alcanza sensibilidad del orden los picomoles y exactitud nunca menor del 10%. Sin embargo, la EM usando MALDI y ESI permite realizar mediciones con sensibilidad (depende del analizador tambien) sub-femtomolar y exactitud de 0.1% y menos de 0.01%, respectivamente (118). En la figura 7 se muestra el espectro ESI del IFN-alpha humano recombinante. Este espectro permitio demostrar la integridad de la proteina recombinante con una exactitud del 0.005%.
La determinacion de la masa molecular con tal exactitud posibilita la deteccion directa de algunas modificaciones en la proteina intacta. CONCLUSIONES La instrumentacion lograda en los nuevos espectrometros de masas, ha colocado a esta tecnica entre las herramientas mas poderosas del quimico, para el estudio de las proteinas naturales y recombinantes. Entre sus rasgos mas caracteristicos estan su alta sensibilidad, alcance de masas, exactitud y amplio espectro de aplicaciones. Su principal limitante esta en que no se dispone de un sistema unico que permita todas las variantes de aplicacion. Por este motivo, en los proximos anos se preve el desarrollo de equipos que combinen las distintas caracteristicas de los instrumentos actuales, tales como: alta resolucion, buena sensibilidad, sencilla manipulacion y posibilidades de acoplamiento con metodos de extraccion/purificacion como HPLC y electroforesis capilar. REFERENCIAS 1. BIEMANN, K.; F. GAPP and J. SEIBL (1959). J. Am. Chem. Soc. 81: 2274. 2. BIEMANN, K. (1972). in Biochemical Aplications of Mass Spectrometry; Walker, G.R., Ed.; John Willey & Sons: New York; 405. 3. BIEMANN, K. (1980). in Biochemical Aplications of Mass Spectrometry, First Supplementary Vol. Walker, G. R.and O.C Dermer, Eds.; John Willey & Sons: New York; 469. 4. BECKEY, H. D. SCHUELTE, (1960). Z. Instrum. 68: 302. 5. SHIMONISHI, Y.; Y-M. HONG; T. MATSUO; I. KATAKUSE; H. MATSUDA (1979). Chem. Lett.: 1369. 6. SHIMONISHI, Y.; Y.-M. HONG; T. KITAGISHI; T. MATSUO; H. MATSUDA, I. KATAKUSE (1980). Eur. J. Biochem. 112: 251. 7. TORGERSON, D.; R. SKOWRONSKI; R. MACFARLANE (1974). Biophys. Res. Comm. 60: 616. 8. SUNDQVIST, B.; I. KAMENSKY; P. HAKANSSON; J. KJELLBERG; M. SALEHPOUR; S. WIDDIYASEKERA; J. FOHLMAN; P. PETERSON; P. ROEPSTORFF (1984). Biomed. Mass Spectrom. 11: 242. 9. MACFARLANE, R. (1982). Acc. Chem. Res. 15: 268. 10. SUNDQVIST, B.; R. MACFARLANE (1985). Mass Spectrom. Rev. 4: 421.6. 11. COTTER, R. (1988). Anal. Chem. 60: 781A. 12. JONSSON, G.; A. HEDIN; P. HAKANSON; B. SUNDQVIST; H. BENNICK; P. ROEPSTORFF (1989). Rapid Commun. Mass Spectrom. 3: 190. 13. JOHNSSON, G.; A. HEDIN; P. HAKANSON; B. SUNDQVIST; B. SAVE; P. NIELSEN; P. ROEPSTORFF; K. JOHANSSEN; I. KAMENSKY; M. LINDBERG (1986). Anal. Chem. 8858: 1084. 14. NIELSEN, P.; P. ROEPSTORFF (1989). Biomed. Environ. Mass Spectrom. 18: 131. 15. TSARBOPOULOS, A.; M. KARAS; K. STRUPAT; B. PRAMANIK; T. NAGABHUSHAN; F. HILLENKAMP (1994). Anal. Chem. 66: 2062. 16. BARBER M.; R. BORDOLI; R. SEDGWICK; A. TYLER (1981). J. Chem. Comm.: 325. 17. FENSELAU, C.; R. COTTER (1982). en IUPAC, Frontiers of Chemistry, Laidler, K., Ed.; Pergamon: New York; 207. 18. SCHRONK, L.; R. COTTER (1986). Biomed. Environ. Mass Spectrom. 13: 395. 19. LEE, T. (1986). en Methods of Protein Microcharacterization, Shively, J., Ed.; Humana Press; New Jersey: 403. 20. BIEMANN, K. (1990). Methods in Enzymology 193: 351. 21. BARBER, M.; R. BORDOLI; G. ELLIOT; R. SEDGWICK; A. TYLER (1983). J. Chem. Soc. Faraday Trans. 79: 1249. 22. DE PAUW, E.; G. PELZER; D. DAO VIET; J. MARIEN (1984). Biochem. Biophys. Res. Commun. 123: 27. 23. BARBER, M.; R. BORDOLI; G. ELLIOT; R. SEDGWICK; A. TYLER (1982). Anal. Chem. 54: 645A. 24. LEVESON, J. (1985). Biomed. Mass Spectrom. 12: 191. 25. BARBER, M.; R. BORDOLI; G. ELLIOT; R. SEDGWICK; A. TYLER; B. GREEN (1982). J. Chem. Soc., Chem. Commun.: 936. 26. LEHMANN, W.; M. KESSLER; W. KONIG (1984). Biomed. Mass Spectrom. 11: 217. 27. WITTEN, J.; M. SCHAFFER; M. O'SHEA; J. COOK; M. HEMLING; K. RINEHART (1984). Biochem. Biophys. Res. Commun. 124: 350. 28. LIGON, W. AND S. DORN (1984). Int. J. Mass Spectrom. and Ion Proc. 57: 75. 29. CLENCH, M.; G. GARNER; D. GORDON; M. BARBER (1985). Biomed. Environ. Mass Spectrom. 12: 355. 30. NAYLOR, S.; F. FINDEIS; B. GIBSON; D. WILLIAMS (1986). J. Am. Chem. Soc. 108: 6359. 31. BIEMANN, K. and S. MARTIN (1987). Mass Spectrom. Rev. 6: 1. 32. MORRIS, H. and F. GREER (1988). Trends in Biotechnol. 6: 140. 33. BIEMANN, K. and H. SCOBLE (1987). Science 237: 992. 34. AITKEN, A. (1990). en Identification of Protein Consensus Sequences, Wiseman, A., Ed., Ellis Horwood Ltd., England; 11. 35. POSTHUMUS, M.; P. KISTAMAKER; H. MEUZELAAR; M. TEN NOEVER DE BRAUW (1978). Anal. Chem. 50: 985. 36. COATES, M. and C. WILKINS (1985). Macromolecules 19: 1255. 37. TAKAYAMA, K.; N. QURESHI; K. HYVER; J. HONOVICH; R. COTTER; P. MASCAGNI; H. SCHNEIDER (1986). J. Biol. Chem. 261: 10624. 38. HILLENKAMP, F. and M. KARAS (1990). Methods in Enzymology 193: 280. 39. KARAS, M.; D. BACHMANN; U. BAHR; F. HILLENKAMP (1987). Int. J. Mass Spectr. Ion Proc. 78: 53. 40. KARAS, M. and F. HILLENKAMP (1988). Anal. Chem. 60: 2299. 41. KARAS, M.; U. BAHR; A. INGENDOH; F. HILLENKAMP (1989). Angew. Chem. 28: 760. 42. BEAVIS, R. and B. CHAIT (1989). Rapid Commun. Mass Spectrom. 3: 233. 43. SPENGLER, B.; D. KIRSCH; R. KAUFMANN; E. JAEGER (1992). Rapid Commun. in Mass Spectrometry 6: 105. 44. STRUPAT, K.; M. KARAS and F. Hillenkamp (1991). Int. J. Mass Spectrom. Ion Process 111: 89. 45. CORNETT, D.; M. DUNCAN and I. Amster (1992). Organic Mass Spectrom. 27: 831. 46. LI, L.; A. WANG and L. Coulson (1993). Anal.Chem. 65: 493. 47. BEAVIS, R. and B. CHAIT (1989). Rapid Commun. Mass Spectrom. 3: 432. 48. BEAVIS, R.; T. CHAUDHARY and B. CHAIT (1992). Organic Mass Spectrom. 27: 156. 49. VORM, O.; P. ROEPSTORFF and M. Mann (1994). Anal. Chem. 66: 3281. 50. MOCK, K.; M. DAVEY and J. COTTRELL (1991). Biochem. Biophys. Res. Comm. 177: 644. 51. NORDHOFF, E.; A. INGENDOH; R. CRAMER; A. OVERBERG; B. STAHL; M. KARAS; F. HILLENKAMP; P.F. CRAIN; (1992). Rapid Commun. in Mass Spectr. 6: 771. 52. ECKERSKORN, C.; K. STRUPAT; M. KARAS; F. HILLENKAMP; F. LOTTSPEICH (1992). Electrophoresis 13: 664. 53. STRUPAT, K.; M. KARAS; F. HILLENKAMP; C. ECKERSKORN; F. LOTTSPEICH (1994). Anal. Chem. 66: 464. 54. VESTLING, M. AND C. FENSELAU (1994). Anal. Chem. 66: 471. 55. FENN, J.; M. MANN; C. MENG; S. WONG; C. WHITEHOUSE (1989). Science 246: 64. 56. LOO, J.; H. UDSETH and R. SMITH (1989). Anal. Biochem. 179: 404. 57. FENG, R. and Y. KONISHI (1992). Anal. Chem. 64: 2090. 58. GREEN, B. and R. OLIVER (1991). Biological Mass Spectrom. 19: 929. 59. CAPRIOLI, R. (1990). Anal. Chem. 62: 477A. 60.ITO, Y.; T. TAKEUCHI; D. ISHI; M. GOTO (1985). J. Chromatogr. 346: 161. 61. CAPRIOLI, R.; W. MOORE; M. MARTIN; B. DAGUE; K. WILSON; S. MORING (1989). J. Chromatogr. 480: 247. 62. MOSELEY, M.; L. DETERDING; J. DE WIT; K. TOMER; R. KENNEDY; N. BRAGG; J. JORGENSON (1989). Anal. Chem. 61: 1577. 63. SUTER, M. and R. CAPRIOLI (1992). J. Am. Soc. Mass Spectrom. 3: 198. 64. MURRAY, K.; T. LEWIS; M. BEESON; D. RUSELL (1994). Anal. Chem. 66: 1601. 65. HUNT, D.; R. HENDERSON; J. SHABANOWITZ; K. SAKAGUCHI; H. MICHEL; N. SERVILIR; L. COX; E. APELLA; V. ENGELHARD (1992). Science 255: 1261. 66. CHOWDHURY, S.; V. KATTA AND B. CHAIT (1990). J. Am. Chem. Soc. 112: 9012. 67. KATTA, V. and B. CHAIT (1991). J. Am. Chem. Soc. 113: 8534. 68. LOO, J.; R. OGORZALEK LOO; H. UDSETH; C. EDMONDS; R. SMITH (1991). Rapid Commun. Mass Spectrom. 5: 101. 69. LE BLANC, J.; D. BEUCHEMIN; K. SIU; R. GUEVREMONT; S. BERMAN (1991). Org. Mass Spectrom. 26: 831. 70. STEVENSON, D.; R. FENG; A. STORER (1990). FEBS Lett. 277: 112. 71. STEVENSON, D.; R. FENG; F. DUMAS; D. GROLEAU; A. MIHOC; A. STORER (1992). Biotechnol. Appl. Biochem. 15: 283. 72. APLIN, R.; J. BALDWIN; C. SCHOFIELD; S. WALEY (1990). FEBS Lett. 277: 212. 73. SHNEIER, A.; C. KLEANTHOUS; R. DEKA; J. COGGINS; C. ABELL (1991). J. Am. Chem. Soc. 113: 9416. 74. MENARD, R.; R. FENG; A. STORER; V. ROBINSON; R. SMITH; A. KRANTZ (1991). FEBS Lett. 295: 27. 75. FENG, R. and Z. YUAN (1990). Proceedings of the 38th ASMS Conference on Mass Spectrometry and Allied Topics, American Society for Mass Spectrometry: East Lansing, MI, 998. 76. GANEM, B.; Y.-T. LI; J. D. HENION (1991). J. Am. Chem. Soc. 113: 6294. 77. GANEM, B.; Y.-T. LI; J. D. HENION (1991). J. Am. Chem. Soc. 113: 7818. 78. BACA, M. and S. KENT (1992). J. Am. Chem. Soc. 114: 3992. 79. BESADA, V.; W. ANTUCH; A. CINZA; I. ROJAS; M. QUINTANA; G. PADRON (1990). Anal. Chim. Acta 239: 301. 80. PADRON, G.; V. BESADA; A. AGRAZ; Y. QUINONES; L. HERRERA; Y. SHIMONISHI; T. TAKAO (1989). Anal. Chim. Acta 223: 361. 81. JENNINGS, K. and G. DOLNIKOWSKI (1990). Methods in Enzymology 193: 37. 82. MCCRERY, D.; T. SACK; M. GROSS (1984). Spectros. Int. J. 3: 57. 83. LEDFORD, E.; S. GHADERI; C. WILKINS; M. GROSS (1980). Adv. Mass Spectrom. 8B: 1707. 84. WHITE, R.; E. LEDFORD; S. GHADERI; C. WILKINS; M. GROSS (1980). Anal. Chem. 52: 1525. 85. CASTORO, J. and C. WILKINS (1993). Anal. Chem. 65: 2621. 86. GROSS, M. (1990). Methods in Enzymology 193: 131. 87. YOST, R. and R. BOYD (1990). Methods in Enzymology 193: 154. 88. BEAN, M.; S. CARR; G. THORNE; M. REILLY; S. GASKELL (1991). Anal.Chem. 63: 1473. 89. MILLINGTON, D. AND J. SMITH (1977). Organic Mass Spectr. 12: 264. 90. BIEMANN, K. (1986). Anal. Chem. 58: 1289A. 91. BIEMANN, K. and S. MARTIN (1987). Mass Spectrom. Reviews 6: 1. 92. HUNT, D.; P. GRIFFIN; J. YATES III; J. SHABANOWITZ; J. FOX; L. BEGGERLY (1989). Techniques in Protein Chemistry, Hugli, T., ed.; Academic: San Diego; 580. 93. JOHNSON, R.; S. MARTIN; K. BIEMANN; J. STULTS; J. WATTSON (1987). Anal. Chem. 59: 2621. 94. BIEMANN, K. (1988). Biomed. Env. Mass Spectr. 16: 99. 95. BORDAS-NAGY; J. DOMINIQUE; D. JENNINGS K. (1992). J. Am. Soc. Mass Spectrometry 3: 502. 96. DOWNARD, K. and K. BIEMANN (1994). J. Am. Soc. Mass Spectrom. 5: 966. 97. BIEMANN, K. (1992). Ann. Rev. Biochem. 61: 977. 98. NAYLOR, S. and G. MONETI (1989). Biomed. Env. Mass Spectr. 18: 405. 99. CODY, R. and B. FREISER (1982). Int. J. Mass Spectrom. Ion Phys. 41: 199. 100. CODY, R. and B. FREISER (1982). Anal. Chem. 54: 1433. 101. BESADA, V.; W. ANTUCH; R. RODRIGUEZ; L. GONZALEZ; G. CHINEA; G. PADRON (1992). Biotecnologia Aplicada 9: 1. 102. MORRIS, H. and P. PUCCI (1985). Biochem. Biophys. Res. Commun. 126: 1122. 103. GARNICK, R.; N. SOLLI; P. PAPA (1988). Anal. Chem. 60: 2546. 104. GRIFFITHS, E. (1994). Biotecnologia Aplicada 11: 1. 105. GEISOW, M. and A. AITKEN (1989). en Protein Sequencing, Findlay, J., Geisow, J., Eds.; IRL Press: Oxfrord; 85. 106. STULTS, J.; HENZEL; W. BOURELL; J. GRIFFIN P. (1990). en Biological Mass Spectrom., Burlingame, A., McCloskey, J., Eds.; Elsevier: Amsterdam; 599. 107. FOOD AND DRUG ADMINISTRATION. Points to consider in the production and testing of new drugs and biologicals produced by recombinant DNA technology (1985). Office of Biologics Research and Review. 108. NARITA, K.; H. MATSUO; T. NAKAJIMA (1975). en Protein Sequence Determination. Needleman, S. B., Ed.; Springer-Verlag: Berlin; 30. 109. MATSUO, H.; Y. FUJIMOTO; T. TATSUNO (1966). Biochem. Biophys. Res. Commun. 22: 69. 110. YARWOOD, A. (1989). en Protein Sequencing. Findlay, J., Geisow, M., Eds.; IRL Press: Oxford; 119. 111. KUMAZAKI, T.; T. NAKAKO; F. ARISAKA; S. ISHII (1986). Proteins: Structure, Function, and Genetics 1: 100. 112. BUCKLER, D.; E. HAAS; H. SCHERAGA (1993). Anal. Biochem. 209: 20. 113. ROSE, K.; L.A. SAVOY; M.G. SIMONA; R.E. OXFORD; P. WINGFIELD (1988). Biochem. J. 250: 253. 114. TAKAO, T.; J. GONZALEZ; H. HORI; K. YOSHIDOME; K. SATO; Y. KAMMEI; Y. SHIMONISHI; presentado en The 40th ASMS conference on Mass Spectrometry and Allied Topics, Washington (Mayo 1992). 115. HENZEL, W.; T. BILLECI; J. STULTS; S. WONG (1993). Proc. Nat. Acad. Sci. USA 90: 5011. 116. PAPPIN, D.; P. HOJRUP; A. BLEASBY (1993). Current Biology 3: 327. 117. MANN, M. and M. WILM (1994). Anal. Chem. 66: 24. 118. CARR, S.; M. HEMLING; M. BEAN; G. ROBERTS (1991). Anal. Chem. 63: 2802. Copyright 1995 Sociedad Iberolatinamericana de Biotecnologia Aplicada a la Salud
The following images related to this document are available:Line drawing images[ba95050d.gif] [ba95050e.gif] [ba95050a.gif] [ba95050c.gif] [ba95050g.gif] [ba95050f.gif] [ba95050b.gif] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
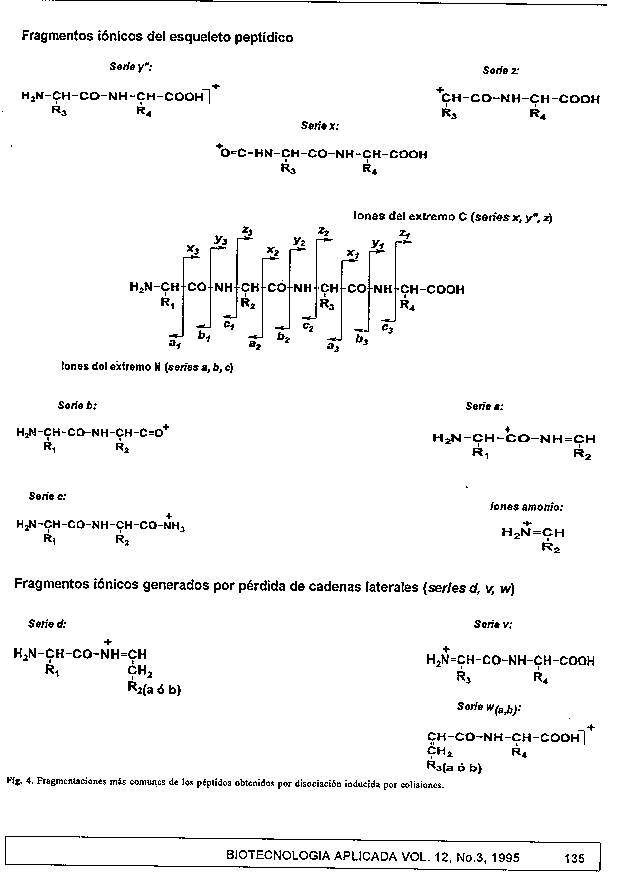{kind=link}
{kind=link}
{kind=link}
{kind=link}