 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
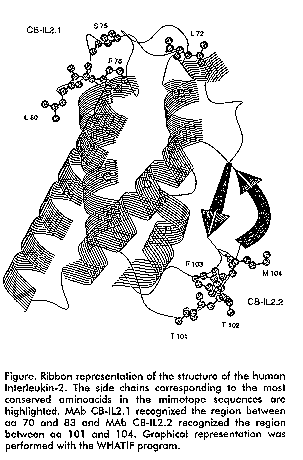
Biotechnologia Aplicada 1998; Vol. 15, No. 2 Trabajos seleccionados del Congreso Biotecnología Habana ´97, diciembre 1-6 1997 Code Number: BA98016 EPITOPIC MAPPING OF TWO ANTI-HUMAN INTERLEUKIN-2 MONOCLONAL ANTIBODIES USING A PHAGE DISPLAYED-PEPTIDE LIBRARY Nelson S Vispo,1 Manuel de J Araña,1 Glay Chinea,1 Ariana G Ojalvo1 and Gianni Cesareni2 1Centro de Ingeniería Genética y Biotecnología, Apartado postal 6162, Ciudad de La Habana CP 10600, Cuba. Introduction Phage displayed-peptide libraries appear to be powerful tools to isolate peptide sequences binding to target molecules. IL-2 has been defined as a class of phylogenetically well-conserved molecule that plays a pivotal role in the immune response regulation. Monoclonal antibodies (MAbs) are central to many areas of current IL-2 /IL-2R research (1). As an integral part of such structure-function studies we are currently cataloguing the binding site locations and detailed specificities of anti-IL-2 MAbs thus maximizing the information available from their use. In order to identify the epitopes recognized by two MAbs (CB-IL2.1 and CB-IL2.2) raised against human Interleukin-2, obtained in our lab, we used a peptide library consisting of nine aminoacids inserted at the N-terminal region of the f1 phage major coat protein pVIII (pVIII-9aa) (2). Materials and Methods Selection of the pVIII-9aa library was performed using the biopanning technique essentially as described by Felici et al. (2). The nucleotide sequences of the gene VIII inserts were determined by single stranded DNA dideoxy sequencing with the chain termination method. Results and Discussion The aminoacid sequences of the inserts were deduced from the nucleotide sequences of the amino terminus of pVIII, and aligned according to their sequence similarity. The epitope recognized by MAb CB-IL2.1 is located in the loop region connecting helix B' with helix C, and involves the beginning of the helix C as well. Among the aminoacids corresponding to the consensus motif L72, S75, K76, L80, R81 and R83 are highly exposed to the solvent. The continuous epitope sequence recognized by MAb CB-IL2.2 was initially identified by the presence of homology to the T101, T102, F103, M104, motif found at the opposite site of the four helix bundle, in a loop that connects helix C with the beta strand Y107-A112 (see Figure). Figure. Ribbon representation of the structure of the human Interleukin-2. The side chains corresponding to the most conserved aminoacids in the mimotope sequences are highlighted. MAb CB-IL2.1 recognized the region between aa 70 and 83 and MAb CB-IL2.2 recognized the region between aa 101 and 104. Graphical representation was performed with the WHATIF program. MAbs directed against native proteins are useful tools for structural and functional studies. Defining the key residue requirement for antibody binding has therefore important implications in the elucidation of relationships between protein structure and function. References
NEW PARADIGMS IN INTERFERON AND CYTOKINE SIGNALING Bryan R and Williams G Department of Cancer Biology Lerner Research Institute, Cleveland Clinic Foundation, Cleveland Ohio, 44195 USA Introduction Interferons (IFNs) are a family of secreted proteins that function as important biological response modifiers. The IFN-genes are transcriptionally induced in response to viral and other microbial infections and IFNs induce rapid activation of gene transcription following binding to specific cell surface receptors. The IFN-stimulated genes (ISGs) modulate the different biological effects of IFNs. The characterization of the molecular mechanisms of IFN-induced gene activation led to the discovery of the Jak-STAT signal transduction pathway. Subsequent studies have shown that many cytokines, growth factors and hormones signal their target cells through this pathway. The Jak-STAT pathway operates through a cascade of specific protein-protein interactions followed by DNA-protein interactions. Protein phosphorylation plays a pivotal role in this process. IFN receptors lacking intrinsic kinase activity are associated with Janus family kinases (Jak) which are activated by receptor engagement. This triggers the activation of the cytoplasmic latent proteins, which are the signal transducers and activators of transcription (STAT), by tyrosine phosphorylation. Activated STATs translocate to the nucleus as oligomeric complexes and interact with the cognate enhancer elements upstream of the ISGs leading to their transcriptional activation and subsequent biological effects. We have recently identifed roles for other molecules in the Jak-STAT pathway. In particular, type I interferon (alpha/beta) treatment causes the phosphorylation and activation of cytosolic phospholipase A2 (cPLA2) which requires JAK1. Moreover, JAK and cPLA2 can be co-precipitated suggesting a close physical interaction. Inhibitors of cPLA2 inhibit IFN alpha/beta induced expression of interferon-stimulated response element (ISRE)-driven genes, but not gamma activated site (GAS)-driven genes indicating that cPLA2 is required for the formation of ISGF3 which binds the ISRE but not STAT1 homodimers which bind GAS elements. We have also implicated the double- stranded RNA (dsRNA) dependent protein kinase (PKR) in signaling by dsRNA, IFNs, TNF-alpha and lipopolysaccharide. PKR is a serine threonine kinase which is induced at a transcriptional level by IFNs and mediates their antiviral activity. PKR is normally inactive but on binding dsRNA it undergoes a conformational change allowing autophosphorylation and dsRNA-independent phosphorylation of substrates. The antiviral activity of PKR is due to autophosphorylation in response to dsRNA followed by phosphorylation of the alpha subunit of initiation factor elF2 resulting in rapid inhibition of translation. In human and mouse cells, dsRNA induction of PKR leads to NFkB activation via the PKR-mediated phosphorylation of IkB. The IFNs, dsRNA, TNF and LPS all fail to activate IRF-1 DNA binding activity in PKR knockout mouse embryo fibroblasts (MEFs) resulting in a selective defect in the induction of genes dependent on IRF-1 (or NFkB). Several genes important in mounting different aspects of host resistance to infection can be classified as wholly or partially dependent on PKR including genes involved in antigen presentation (class I MHC), chemotaxis (the chemokines IP-10, Myg, JE, Rantes) antimicrobial activity (iNOS) and apoptosis (FAS). The induction of the cell adhesion molecules VCAM and E-selectin by dsRNA is also mediated via a PKR-dependent pathway. The mechanisms of direct activation of PKR by cytokines remain to be determined. Whether this occurs through Jak-dependent mechanisms or other signals generated by receptor engagement is not known. Direct interactions of PKR with components of the Jak-STAT pathway have been identified. There is deficient STAT3 binding to the GAS element in the c-fos promoter in extracts from PKR knockout MEFs compared to wild type cells in response to PDGF, although the response of STAT3 to other stimuli remains unaffected. PDGF induces a rapid and transient increase in the interaction between PKR and STAT3. While the mitogenic effect of PDGF on PKR wild type and knockout cells is identical, c-fos gene induction by PDGF is deficient. Therefore the mitogenic pathway activated by PDGF is PKR independent and may be distinguished from the PKR dependent pathway required for immediate early gene expression which involves a PKR-STAT3 interaction. Physiological levels of PKR are required for mediating an apoptotic response to different stimuli including dsRNA. PKR knockout MEFs are resistant to apoptotic cell death in response to dsRNA, TNF-alpha, or LPS. The mechanism for the suppression of apoptosis in PKR knockout cells was linked to a defect in the activation of the DNA- binding activity of the transcription factor, IFN Regulatory Factor-1 (IRF-1) in response to dsRNA, LPS or TNF-alpha. These results reveal an unexpected role for normal cellular basal levels of PKR in mediating several forms of stress-induced apoptosis through regulation of lRF-1 activity. PKR does not appear to be involved in mediating apoptosis required for normal growth and development as the PKR knockout mice have a normal life span and there are no gross abnormalities in peripheral lymphoid compartments. However, while Fas mRNA expression is strongly upregulated in wildtype cells by dsRNA and LPS, this induction, is much reduced in PKR knockout MEFs. The induction of Fas on wildtype MEFs by dsRNA results in cells becoming sensitive to killing by an antagonist anti-Fas antibody, Jo2, which induces apoptosis specifically by stimulating the Fas receptor. PKR knockout MEFs treated with dsRNA remain insensitive to killing by the Jo2 antibody. Thus, PKR may play a role in other processes where up-regulation of Fas is required for activating apoptotic pathways to control cellular stress responses.
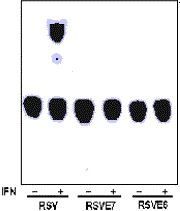
MOLECULAR BASES FOR HUMAN PAPILLOMAVIRUS RESISTANCE TO INTERFERON ACTION Silvio E Perea, Omar López-Ocejo, Gustavo Bracho, Rolando García-Milián and Manuel de J Araña División de Farmacéuticos, Centro de Ingeniería Genética y Biotecnología, Apartado postal 6162. Ciudad de La Habana CP 10600. Cuba Introduction Human Papillomavirus (HPV) is mostly associated with the development of cervical carcinoma (1). The two major transforming proteins, E6 and E7, a re generally conserved after viral integration into the cell genome and their expression is necessary for maintaining the malignant cell phenotype. Both viral oncoproteins respectively complex with products from p53 and Rb genes and such events appear to be determinant for cellular transformation produced by these DNA tumor viruses. IFNs are a family of biological response modifiers that exhibit antiviral, antiproliferative and immunomodulating functions. Binding of IFNs to their receptors triggers the assembly of a cytoplasmic protein complex and its translocation to the nucleus, where the activated complex can promote transcription by binding to ISREs (2). In this work, we examined the effect of E7/E6 oncoproteins on the ISRE activation by IFN and investigated the functional domain of HPV-16 E7 engaged with the interference elicited by such oncoprotein. We found that E7 phosphorylation site by Casein Kinase II (CK II) was essential for inhibiting the ISRE activation by IFN suggesting that this molecular event is involved on the resistance of oncogenic HPVs to IFN action. Materials and Methods Cells Caski (human cervix epidermoid carcinoma, HPV-16 positive), HeLa (human cervix adenocarcinoma, HPV-18 positive), RHEK-1 (human epidermic keratinocytes), Hep-2 (human laryngeal carcinoma). Plasmids RSV E6/E7 (E6 or E7 ORFs driven by RSV enhancer/promoter) p24Gly (Rb-binding E7 mutant), p31/32Arg/Pro (E7 phosphorylation mutant), p566Pro (E7 transformation mutant), 3X ISRE CAT (IRF-1 binding site from fused to CAT gene) Transient transfection experiments Cells were transfected by DEAE-dextran method with 3XISRE-CAT vector and constructs expressing either HPV-16 E7 mutants or wild type. Results and Discussion We demonstrated that HPV-16 E7 and E6 oncoproteins largely abrogated the activation of a GBP ISRE reporter by IFN suggesting that both viral oncoproteins seem to impair the IRFs/Stats function (Figure 1). These findings are in line with other ones observed in patients where high levels of E7 oncoprotein were correlated with the lack of IFN response in terms of ISGs activation and antiproliferative effect. Figure 1. Effect of the E7 and E5 expression of HPV 16 on the activation of the ISRE sequence. We further investigated the HPV-16 E7 functional domain engaged with the impairment of the ISRE activation by IFN. We found that substitution of Ser 31/32 by non-phophorylitable residues on E7, fails to abrogate the ISRE reporter activation by IFN (Figure 2). In other approach using heparin as CK II inhibitor we observed an increase of the IFN-induced GBP mRNA levels. In CaSki cells, heparin pre-treatment increased the sensitivity of these cells to the antiproliferative effect of IFN. Figure 2. Analysis of the E7 functional domain envolved in the inhibitory effect of the activition of the ISRE sequence. Taking together these results, we suggest that HPV resistance to IFN action is in part mediated by E7 phosphorylation event. Likewise, such results could lead to the speculation that CK II inhibition would be therapeutically useful in treating HPV-patients resistant to IFN. Referencias
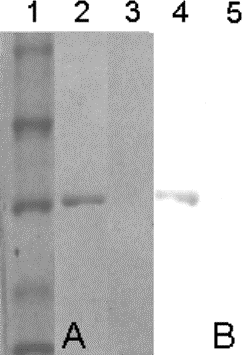
CLONING AND EXPRESSION OF AN ANTI CEA SINGLE CHAIN ANTIBODY FRAGMENT IN THE METHYLOTROPHIC YEAST Pichia pastoris Freya M Freyre, Javier E Vázquez, Marta Ayala, Leonardo Canaán-Haden, Hanssel Bell, Alberto Cintado and Jorge V Gavilondo Centro de Ingeniería Genética y Biotecnología, División de Inmunotecnología y Diagnóstico, apartado postal 6162, Ciudad de La Habana, 10600, Cuba. E-mail: Jorge.Gavilondo@cigb.edu.cu Introduction Single chain Fv (scFv) antibody fragments are genetically engineered recombinant fusion proteins in which the variable light (VL) and variable heavy (VH) chain domains are connected by an artificial linker (1). The major reason for which heterologous production of scFv and its derivatives forms is a field in rapid development is the potential application of these small antigen binding proteins if produced at a low cost. Such applications include areas where the specific binding of the antibody fragment is useful, as in tumor therapy and diagnostics, in affinity purification, and as biocatalysts (2, 3). Here we report the cloning and expression of an active scFv antibody fragment (scFvCEA1904his6) specific to Carcinoembryonic Antigen (CEA) in the extracellular medium of the methylotrophic yeast Pichia pastoris. The exploitation of P. pastoris as a tool to produce useful quantities of biologically relevant recombinant proteins has gained special attention due to the existence of well-established fermentation methods and of expression plasmids with very powerful and efficiently methanol-regulated promoters (4). Experimental procedures The anti-CEA scFv encoding gene was excised from a bacterial vector in which it was originally cloned (5), and inserted into the P. pastoris expression vectors pHIL-S1 (Invitrogen, San Diego, CA) and pPS7 (CIGB, Havana, Cuba), bearing the Pichia acid phosphatase 1 (PHO.1) and Saccharmomyces cerevisiae sucrose invertase 2 (Suc 2) signal sequences, respectively, and the methanol-inducible alcohol oxidase 1 (AOX.1) promoter. These constructs were denominated pPSCEA1904his6 and pPSChis6CEA. Competent P. pastoris GS115 his4(mut+) cells (Invitrogen) were electroporated by application of an exponential decay wave electric pulse of 12 kV/cm, for 4.6 ms, in the presence of BgIII-linealized recombinant construct (pPSCEA1904his6). Several HIS+ transformants were analyzed for the presence of the gene product of interest in the extracellular medium and in the cellular pellet. Proteins were detected in SDS-PAGE gels stained with Coomassie brilliant blue, and after electrotransfer to nitrocellulose, using specific rabbit anti scFv polyclonal antibodies, and HRPO conjugated-anti-rabbit Igs. The biological activity of scFv was monitored with a specific ELISA, where the CEA was coated to the solid phase of 96 microtiter plate wells followed by application of the two-fold diluted samples of culture supernatant before and after induction. Culture supernatant of an induced Human serum albumin (HAS) producer GS115 strain (Invitrogen) as well as from GS115 strain transformed with pHIL-S1 were used as negative controls. Anti scFv-CEA rabbit Igs were used as revealing reagent. Results and Discussion The anti CEA scFv fragment had been initially expressed in E. coli using several strategies (5, 6) but its yields were limited by a low level of production of the protein to the periplasm or by the inefficiency inherent to refolding of denatured inclusion bodies. To overcome these limitations, the scFv encoding gene was cloned into the P. pastoris vectors pHIL-S1 and pPS7 for expression as secreted protein. Two distinct phenotypes among 13 GS115/pPSCEA1904 His+ transformants analyzed were obtained: clones exhibiting slow growth in methanol (muts), and clones capable of still utilizing methanol (mut+). These phenotypic differences were presumed to be due to the interruption of the AOX 1 gene by insertion of the expression plasmid in the case of the muts phenotype, and integration of the expression plasmid at an alternative site in the case of the mut+ phenotype. Only one clone of each phenotype group secreted proteins of the expected size (27.5 kDa) into the culture medium at high levels (100-200 mg/L). This protein was identified as the scFv by SDS-PAGE and Western-blot. The secreted anti-CEA scFv recombinant protein comprises the vast majority of the total protein in the culture medium, which serves as a first step in purification of the expressed protein (Figure 1). It is a specific advantage of secretion in P. pastoris, since the organism secretes only very low levels of native proteins (4). The secreted scFv also binds to CEA in a direct ELISA even when the sample of culture medium is two-fold diluted. These results suggest that secretion of functional scFv fragments by P. pastoris can provide a low cost, high yield alternative to current bacterial scFv expression systems. Figure 1. Electrophoretic (A) and Western-blot (B) detection of secreted anti-CEA scFv in the culture medium of a P. pastoris mut+GS115/HIS+ transformant. References
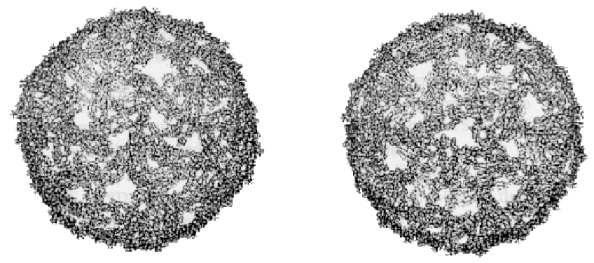
VISUALIZACIÓN MEDIANTE EL MICROSCOPIO DE FUERZAS ATÓMICAS DEL CONECTOR DEL BACTERIÓFAGO F 29 SUMERGIDO EN SOLUCIÓNVélez M,1 Carrascosa JL,2 Müller DJ3,4 y Engel A3 1Laboratorio de Bajas Temperaturas C-III, Departamento Física de la Materia Condensada y Introducción EI ensamblaje de los virus bacteriófagos requiere que los distintos componentes estructurales del mismo interaccionen de forma secuencial y específica. En este complejo proceso, las proteínas que forman la región que conecta la cabeza con la cola del virus (conectores) participan activamente en el ensamblaje de las procabezas y, posteriormente, en el reconocimiento y empaquetamiento del DNA viral dentro de la cabeza del fago (1). La microscopia electrónica ha permitido obtener amplia información estructural sobre el conector del bacteriófago F 29 (2, 3). Sin embargo, las limitaciones técnicas de las reconstrucciones tridimensionales obtenidas por microscopia eléctronica no han permitido obtener información detallada sobre la topografía de la superficie del conector, a pesar de la importancia que esta información tendría para entender el proceso de interacción con DNA y los otros ligandos que participan en el proceso de reconocimiento y empaquetamiento de DNA. El microscopio de fuerzas atómicas operando en líquidos ha demostrado recientemente ser capaz de proporcionar imágenes topográficas con resolución subnanométrica de proteínas de membrana (4). Hemos utilizado esta técnica para estudiar la superficie de cristales bidimensionales de conectores de F 29 adsorbidos sobre mica. La nueva información estructural adquirida complementa la obtenida por crioelectromicroscopía. Resultados Se tomaron imágenes de la superficie de cristales bidimensionales de conectores adsorbidos firmemente sobre la superficie de mica y sumergidos en solución tampón (5). En la imagen presentada en la Figura 1 pueden observarse los detalles estructurales de la superficie de cada conector. En las imágenes tomadas aplicando menor fuerza (Figura 1a) se distinguen claramente los extremos estrechos de los conectores sobresaliendo de la superficie del cristal. Sólo pueden observarse los detalles estructurales del extremo más ancho cuando imperfecciones del cristal (círculo blanco en la Figura 1a) permiten que la punta penetre hasta ellos. Si se aplica con la punta mayor fuerza se desplazan reversiblemente los extremos estrechos de los conectores y se distinguen entonces las doce subunidades y el orificio central de la base más ancha (Figura 1b). Figura 1. Topografía con alta resolución de la superficie de un cristal bidimensional de conectores de F 29. Conectores visualizados aplicando una fuerza de 50-100 pN (a) y de 150 pN (b). Las barras equivalen a 50 nm. La escala de grises abarca un rango de 4 nm . Las imágenes obtenidas con microscopía de fuerzas confirman la localización de dos conectores por celda unidad orientados en direcciones opuestas, ta1 y como lo indica el grupo p4212 descrito anteriormente (3). Permiten también determinar que la altura mínima del conector es de 7,6 ± 0,5 nm y estimar que la altura del extremo estrecho es de 5,0 ± 0,6 nm mientras que la del extremo más ancho es de 2,6 ± 0,6 nm. Se observa también que la forma del canal no es cilíndrica sino tronco-cónica. Otra información relevante obtenida es la vorticidad que presenta la superficie de las doce subunidades del extremo ancho del conector. Estos datos contribuyen a refinar el modelo tridimensional del conector existente (Figura 2c). Figura 2. a) Vista en perspectiva de la imágen promediada y simetrizada de la topografía del cristal del F 29. La topografia se obtuvo a partir de promedios simetrizados del extremo estrecho y del extremo ancho del conector. La celda unidad (señalada por las cuatro esquinas blancas) mide 16,5 nm x 16,5 nm. b) Vista lateral de la reconstrucción tridimensional del conector obtenida a partir de imágenes inclinadas de cristales teñidos negativamente (adaptado de (2)) c) Vista lateral del modelo tridimensional obtenidos de los datos en b) después de combinar con la información obtenida con el microscopio de fuerzas atómicas. Referencias
CHARACTERIZATION AND 3D MODEL OF A NEW PROTEINASE INHIBITOR ISOLATED FROM Stichodactyla helianthus Vivian Huerta,1 Vivian Morera,1 Nelia López,1 Lázaro Betancourt,1 Vladimir Besada,1 Gabriel Padrón,1 Gipsi Lima,2 María de los A Chávez,2 Julieta Delfín2 and Joaquín Díaz2 1División de Química-Física, Centro de Ingeniería Genética y Biotecnología. Apartado postal 6162, Ciudad de La Habana, CP 10600, Cuba. E-mail: vivian.morera@cigb.edu.cu Introduction Protein proteinase inhibitors are widely distributed in living organisms and classified upon their similarities according to sequence, topology, active site localization and the binding mechanism. Proteinase inhibitors from sea anemones are specially interesting as they represent the phylogenetically oldest aprotinin type inhibitors known until now. They can be easily isolated in large quantities. Isoinhibitors with different inhibitory specificity may provide desirable properties for therapeutic uses and for the study of structure-functions relationships. In 1996 we reported the isolation and characterization of the ShPI-I protease inhibitor from the sea anemone Stichodactyla helianthus (1). Here we report the primary structure and the dissociation constant against trypsin of a new proteinase inhibitor ShPI-2, isolated from the same source. We also discuss a 3D model of the ShPI-2/Trypsin complex. Results and Discussion ShPI-2 was isolated from the whole body of the anemone as described (1). A fraction with protease inhibitory activity was subjected to a final purification step by rp-HPLC using a C8 column. ShPI-2 constituted the main fraction and was obtained with high purity. An average molecular mass of 6196.0 Da. was obtained by FAB-MS on a Jeol JMS-HX110 mass spectrometer. 52 N-terminal residues were determined by automatic sequencing of S-carboxamidomethylated protein using a Knauer 810 dual-phase sequencer. The remaining sequence including the C-terminus was established by digestion of the protein with endoproteinase Glu-C and combining automatic sequencing and FAB-MS data. FAB-MS analysis of peptides obtained by successive digestions with endoproteinase Glu-C and Lys-C corroborated the sequence (Figure 1). Figure 1. ShPI-2 amino acid sequence. In bold faces are denoted residues that belong to the sequence common pattern of the BPTI Kunitz family. The observed and expected (in parentheses) mass values are shown for each peptide. ShPI-2 has 92 % of sequence identity with ShPI-I from S. helianthus. Disulfide bond between Cys3-Cys53 was determined by partial acid hydrolysis followed by successive digestions with pepsin and trypsin. Sequence similarity searches revealed that ShPI-2 has the sequence pattern common to the Kunitz family of BPTI (Figure 1). Due to this fact disulfide bridges between Cys12-Cys36 and Cys28-Cys49 were assigned by homology with the inhibitor protein family. ShPI-2 has a strong inhibitory capacity against trypsin (Ki 3.8 x 10-10) determined according to Bieth (2). The 3D model of the ShPI-2/Trypsin complex was build using the WHATIF program (3). The structure of the BPTI/Trypsin complex (2PTC PDB code) was used as template for the regions that interact with trypsin and the structure of ShPI-I solved by RMN (1SPH PDB code), for the rest of the inhibitor. The most important interactions for the trypsin inhibition are present in our model as well as in several structures of enzymes/inhibitors complexes. There are specific residues in the contact region with trypsin that could explain the difference in the inhibition constant between ShPI-2 and BPTI. The fact that the differences between ShPI-I and ShPI-2 sequences are in regions that are not in contact with trypsin explains the similarity in their inhibition constants (ShPI-I, Ki 1.1 x 10-10). References
MODELING THE FLAVIVIRUS ENVELOPE. PREDICTION OF FUNCTIONAL RESIDUES BY THE ANALYSIS OF E-GLYCOPROTEIN SEQUENCE CONSERVATION PATTERNS Glay Chinea, Tirso Pons, Osvaldo Olmea and Gabriel Márquez Division of Physical Chemistry, Center for Genetic Engineering and Biotechnology. PO Box 6162, Havana, Cuba. E-mail: Protein.Design@cigb.edu.cu Introduction The glycoprotein E is the major protein of the proteolipid envelope of flaviviruses. It mediates the interaction with cellular receptors and the acid catalized membrane fusion, constituting an important factor determining tropism, host range, virulence and protective immunity. Recently, the X-ray crystal structure of a soluble fragment of protein E from tick born encephalitis virus (TBE) has been solved, showing that the protein forms head-to-tail homodimers which apparently lay parallel to the viral membrane (1). Biochemical evidences suggest that these dimers are organized in a yet undefined network-like structure. The functional sites of protein E have not been mapped neither, though some speculations have been made concerning the location of receptor binding sites and the fusion peptide. Here we report the modeling of a symmetrical lattice structure that could be adopted by the protein E dimers in the surface of the virions and which is consistent with the experimental data. Furthermore, we have analyzed the sequence conservation patterns of flavivirus E proteins, suggesting the location of likely functional residues. Materials and Methods Modeling of the viral envelope and homology modeling of dengue virus E protein was achieved using the program WHATIF (2). Sequences of flavivirus E proteins were obtained from SWISSPROT, Genbank and EMBL databases. Secondary structure predictions were made with PHD (3). Prediction of functional residues were carried out by Sequence Space Analysis (4). Results and Discussion We have built a model of the viral envelope consisting of an icosahedral T=3 lattice of protein E monomers. Modeling was accomplished according to the following assumptions: virus diameter is 500 Å, the C2 symmetry axis of protein E crystal dimers coincide with the icosahedral pseudo- and real C2 symmetry axis, bumps between protein atoms are forbidden and the distance between the C-terminus (residue 395 of TBE) of each subunit and the icosahedral pseudo C3 symmetry axis is minimal. The modeled structure is shown in Figure 1. The resulting particle is formed by 90 dimers. The space around the pseudo-C3 symmetry axis could be occupied by the C-terminal portions of the ectodomain which links the crystallized fragment with the membrane anchor. This segment is predicted to be constituted of two amphipathic helixes separated by a highly conserved loop and it could play an important role in the pH induced oligomeric transition from dimers to trimers, which seems to be necessary for the fusogenic activity. The conservation patterns present in flavivirus E proteins should reflect the evolutionary constrains imposed by the biological function. Flavivirus conserved residues are mostly buried or exposed into the inner surface. Three major clusters were apparent: the tip of domain II, likely to be important for fusion activity; the putative loop at the C-terminal fragment of the ectodomain, whose probable role was discussed above; and an ionic cluster located in the interface between domain I and III, which we predict to participate in the low pH triggered oligomeric transition, stabilizing indirectly the dimers at neutral pH and destabilizing it at acidic pH. The outer surface of the protein is highly variable among flavivirus complexes and types. Further analysis of the dengue virus complex showed a conserved residue cluster located at the upper lateral surface of domain III, pointing toward the C5 and C3 symmetry axis of the modeled virions. We believe it could be a receptor binding site, involved in the virus interaction with a receptor from human and/or mosquito cells. Figure 1. Stereo view of the modeled icosahedral envelope of TBE. References
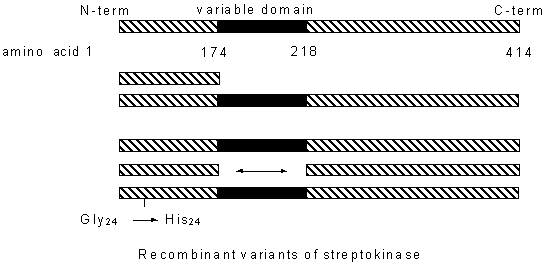
STREPTOKINASE-MEDIATED PLASMINOGEN ACTIVATION: MOLECULAR STUDIES USING GENETICALLY ENGINEERED STREPTOKINASE VARIANTS Sergio Lizano Instituto Clodomiro Picado, Facultad de Microbiología, and Departamento de Bioquímica, Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica Introduction Streptokinase triggers a non-proteolytic activation of plasminogen by forming a stoichiometric complex with plasminogen (1), which develops proteolytic activity after complex formation. A molecular approach was applied to address this question by studying the formation of streptokinase-plasminogen complexes on a solid phase and by constructing genetic variants of the streptokinase molecule using recombinant DNA technology (see Figure). Figure: Recombinant variants of streptokinase Materials and Methods The following streptococcal strains were chosen: NZ 131 (group A Streptococcus pyogenes, M-type 49), SP 13013 (group A S. pyogenes, M type 1), and H46A (group C S. equisimilis). Streptokinase sequences were amplified by polymerase chain reaction (PCR) followed by ligation to pUC18 and subcloning to expression vector pGEX-3X in translational frame with the glutathione-S-transferase gene (2). Transformed colonies were selected and GST-streptokinase was expressed as described by Lizano and Johnston (3). An internal polymorphic region of the streptokinase molecule implicated in the pathogenesis of glomerulonephritis (4) was deleted and replaced with a double stranded linker constructed by annealing the following oligonucleotides designed to maintain the reading frame upon ligation to the linearized pGEX-3X containing the remainder of the streptokinase gene. Site directed mutagenesis was performed by Unique Site Elimination (U.S.E.) mutagenesis according to Deng and Nickoloff (5). Human Glu-plasminogen was purified according to Deutsch and Mertz (6). The recombinant constructs were assayed according to Kulisek et al. (7). Results and Discussion Streptokinase immobilized on affinity matrices via its NH2-terminal fusion to GST or by incorporation of a COOH-terminal poly-histidine "tail" formed active, non-fragmented complexes with plasminogen; this provided an alternative approach to pre-proteolysis plasminogen activation (3). The N- and the C-terminal conserved domains bind plasminogen independently, yet both must be simultaneously present to achieve a fully active complex with plasminogen. Moreover, mutagenesis studies of glycine 24 of streptokinase previously reported to be indispensable for activity indicated that this residue is rather non-essential for activation. Future efforts to characterize the structure/function relationship of streptokinase may influence the engineering of streptokinase to improve its therapeutic potential and explain its role in streptococcal disease. References
Copyright 1998 Elfos Scientiae The following images related to this document are available:Line drawing images[ba98016a.gif] [ba98016d.gif] [ba98016h.gif] [ba98016g.gif] [ba98016e.gif] [ba98016b.gif] [ba98016f.gif] [ba98016c.gif] [ba98016i.gif] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
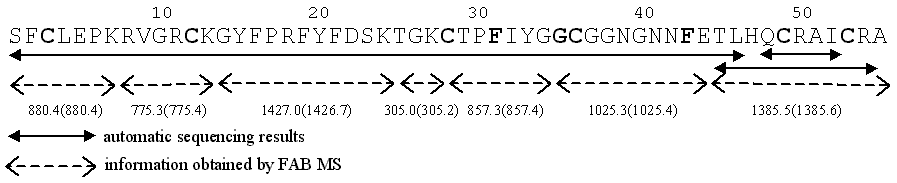{kind=link}
{kind=link}
{kind=link}