 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Biotecnología Aplicada 1998;15:219-226 CORRECEPTORES DEL VIRUS DE LA INMUNODEFICIENCIA HUMANA TIPO 1 Enrique Iglesias División de Vacunas, Centro de Ingeniería Genética y
Biotecnología. apartado postal 6162,
Code Number: BA98035
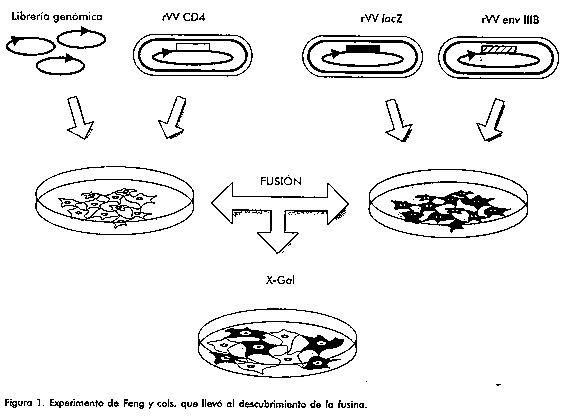
ABSTRACT For a long time ago is known that CD4 molecule is not enough to promote viral entry. A new period in the viral studies began when the first correceptor was reported in May 1996. The 5% of the people resistant to HIV infection can be explained by the knowledge of the genetic polymorphism of one of them (CCR5). Without any doubt their inclusion in HIV-1 pathogenesis models make possible to predict in a more accurate way the chain of phenomena associated with infection. Although it is possible that new therapeutic strategies are generated based on the selective blockade of the second HIV-1 receptors. Keywords: co-receptor, CCR2b, CCR3, CCR5, CXCR4, HIV RESUMEN Desde mucho tiempo atrás se conocía que la molécula CD4 no era suficiente para promover la entrada viral. Cuando en mayo del año 1996 se reportó el descubrimiento del primer correceptor del VIH-1 se abrió una nueva etapa en los estudios del virus. El conocimiento del polimorfismo genético asociado a uno de ellos (CCR5) permite explicar el 5% de los casos de personas que poseen una gran resistencia a la infección. Sin lugar a dudas su incorporación en los modelos de patogénesis viral permite explicar y predecir más cabalmente la sucesión de fenómenos asociados a la infección. Además es posible que se generen nuevas estrategias terapéuticas basadas en el bloqueo selectivo de los segundos receptores del VIH-1. Palabras claves: correceptor, CCR2b, CCR3, CCR5, CXCR4, VIH Introducción La patogénesis del virus de la inmunodeficiencia humana tipo 1 (VIH-1) fue primariamente relacionada con un receptor viral a partir de observarse que las células T CD4+ eran depletadas selectivamente en pacientes con síndrome de inmunodeficiencia adquirida (SIDA) [1]. Un tiempo después se obtuvo la inmunoprecipitación con un anticuerpo monoclonal (AcM) anti-CD4 de la glicoproteína 120 (gp120) unida a CD4 a partir de incubar virus marcado radiactivamente con células CD4+ [2]. Esto indicó que la interacción primaria de la envoltura viral era con la molécula CD4 de la superficie celular. Sin embargo, la posibilidad de que un segundo receptor estuviese involucrado en la fusión del virus se infería de una serie de observaciones. Primero, las cepas del VIH-1 están restringidas en su tropismo: algunas prefieren los linfocitos (linfocitotrópicas), mientras otras los monocitos/macrófagos (monocitotrópicas) a pesar de que ambas células expresan CD4 a niveles comparables [3-6]. Por consiguiente, es de suponer que otra u otras moléculas estén involucradas para generar alguna diferencia. Segundo, la transfección de líneas celulares humanas no linfoides con CD4 permite la replicación viral del VIH-1; pero la transfección de líneas murinas con construcciones para la expresión de CD4 no es suficiente para permitir la replicación del virus [7]. Además células no humanas que expresen CD4 son permisivas para la infección por el VIH-1 cuando se funden con membranas humanas [8] o se logra la formación de un heterocarionte transitorio [9, 10]. Se han aportado algunas evidencias sobre la existencia de otros receptores además de CD4. Así en un estudio se observó que líneas de células epiteliales de colon, que no expresan CD4, eran infectadas por el VIH-1 a través de otro receptor: un glicolípido neutral nombrado galactosilceramida [11]. Este glicolípido también se ha hallado en neuronas y se ha sugerido que podría actuar como receptor independiente de CD4 para el VIH [12]. Sin embargo, la entrada a través de la galactosilceramida es muy ineficiente. También receptores de la fracción constante (Fc) y el complemento han sido implicados como receptores del virus [13]. Durante los años precedentes varias proteínas presentes en las membranas de las células humanas han sido propuestas como correceptores del VIH-1. Entre estas están: el receptor de adhesión de leucocitos (LFA) [14], una serin-proteasa de monocitos [15], CD26 [16], CD7 [17] y CD44 [18]. Sin embargo, en experimentos tendientes a probar su necesidad para lograr la entrada viral ninguna ha sido eficiente. Recientemente, la búsqueda del correceptor para el VIH-1 ha convergido con el estudio de un grupo de citoquinas, nombradas particularmente quimoquinas, y sus receptores. Las quimoquinas actúan fundamentalmente como quimoatrayentes induciendo la migración de leucocitos a los sitios de inflamación aunque, además, están involucradas en otras funciones inmunes [19]. Ellas se caracterizan por poseer cuatro residuos conservados de cisteínas. Cuando los primeros dos residuos están separados por algún otro aminoácido (aa) se clasifican como CXC o a-quimoquinas, cuando están adyacentes como CC o b-quimoquinas. Se ha descrito una quimoquina nombrada linfotactina que sólo tiene dos de los cuatro residuos de cisteínas, por lo cual se separan en una tercera familia C de la cual es el único miembro hasta el momento [20]. Los receptores de las quimoquinas se nombran de acuerdo a la clasificación de su ligando como CXCR (la R por receptor) o CCR, y se agrega un número indicativo del orden en que se han ido caracterizando. Se ha demostrado que algunos receptores de quimoquinas actúan como segundos receptores para la entrada del VIH-1 a las células. Primer correceptor descubierto: "fusina" o CXCR4 Fusina fue el nombre propuesto para el primer correceptor descubierto, reportado en mayo de 1996 [21]. Sus descubridores, Feng y cols., primero transfectaron células NIH 3T3 con una biblioteca genómica humana hecha de la línea HeLa y después infectaron éstas con un virus vaccinia recombinante para el marcador CD4. Luego, estos transfectantes fueron mezclados con células NIH 3T3 coinfectadas con vaccinias recombinantes para los genes lacZ y el gen env codificante para la envoltura del aislamiento VIH-1 IIIB. Finalmente, terminada la incubación tiñeron in situ con x-Gal y contaron las células fusionadas teñidas (Figura 1). Obtuvieron consistentemente más células teñidas cuando transfectaban con toda la biblioteca que cuando lo hacían con un solo plasmidio escogido, aleatoriamente, de la biblioteca. Después de varios fraccionamientos y ensayos de actividad b-Gal lograron aislar clones individuales capaces de promover la fusión de células que expresaban CD4 con las que expresaban la envoltura del virus. Se secuenció un fragmento de ADN complementario de 1,659 kb y se verificó que el mayor marco abierto de lectura, codificante para una proteína de 352 aa, se correspondía con un gen previamente reportado por varios grupos. Nadie había hallado función alguna para esta proteína, ni la habían relacionado con el VIH; esa fue la causa por la que propusieran llamarla fusina. Figura 1. Experimento de Feng y cols. que llevó al descubrimiento de la fusina. Los autores del referido artículo llegaron a varias conclusiones a partir de una serie de experimentos. En primer término, la expresión de la fusina tanto en células no humanas como humanas (que expresen CD4, de otro modo no hay infección) las hace susceptibles a la infección por el VIH-1. En segundo lugar, la fusina es eficiente promoviendo la entrada a las células CD4+ solamente cuando la infección es ensayada con aislamientos de VIH linfocitotrópicos; no es así para los monocitotrópicos. De ahí que postularan que la fusina es el correceptor mediante el cual las cepas linfocitotrópicas del VIH-1 entran a células que expresan el marcador CD4. Muchos grupos han continuado investigando la capacidad de CXCR4 de actuar como correceptor. Por ejemplo, a principios del año pasado se publicó una comunicación rápida que mostraba que la expresión de la fusina en una línea CD4+ humana, resistente a la infección, la volvía susceptible a la infección por VIH-1 y VIH-2 [22]. La fusina era referida anteriormente con otros nombres: L5 [23], hFB22 [24], pBE1.3 [25], HUMSTR [26], HM89 [27] y LESTR [28]. La convención actual de nomenclatura ha nombrado a la fusina como CXCR4 [29, 30]. Su función como receptor del SDF-1 (stromal cell derived factor 1) fue hallada poco tiempo después de su vinculación con el VIH [29, 30]. Al ser el SDF-1 una quimoquina CXC, queda entonces definida la fusina como un receptor de quimoquinas. CXCR-4, a semejanza del resto de los receptores de quimoquinas, es una proteína G, generadora de influjo de Ca2+, que atraviesa siete veces la membrana plasmática [31]. La interacción virus/CXCR4 se ha estudiado con
profundidad. Aprovechando que entre el CXCR4 de rata y el humano existe más de un
90% de identidad, y que las principales diferencias se ubican en los dominios extracelulares, se
han producido diversas proteínas quiméricas rata/humano y mutantes de
deleción de CXCR4 [32]. Estas proteínas se expresaron en una línea
de glioma humano CD4+, y en ella se ensayó la capacidad infectiva de tres
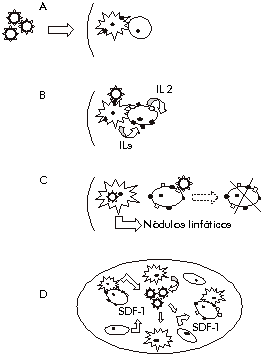
aislamientos de VIH-1 (LAI, NDK, NL4.3) y uno de VIH Con el uso de una línea neuronal CD4- se ha evidenciado infección del VIH-1 mediada por CXCR4 solamente [34], sin participación de la galactosilceramida como había reportado otro grupo [12]. Lo anterior coincide con un reporte similar para el VIH-2 [35]. Estos estudios muestran que la fusina puede actuar a su vez como receptor. CCR5 Tan solo un mes después del reporte sobre CXCR4 se reportaba por cinco grupos independientes el segundo correceptor -CCR5- [36-40]. Uno de estos grupos fue el mismo que reportó la fusina. En todos los casos los autores fueron a la búsqueda del receptor para las quimoquinas RANTES, MIP-1a y MIP-1b, teniendo como premisa los trabajos de Gallo y Lusso, hacía un año, quienes mostraron que éstas impedían la replicación viral [41]. A lo anterior se unía el hecho de que el primer correceptor reportado era un receptor de quimoquinas. Así, todo indicaba claramente que la actividad antiviral de las quimoquinas se podía deber a un bloqueo de la entrada del virus a la célula. El grupo de Berger usó nuevamente virus vaccinia recombinantes para lacZ y env (de varios aislamientos) para ensayar la fusión con células de sangre periférica activadas con fitohemaglutinina (PHA) en presencia de las diferentes quimoquinas [36]. En este experimento se comprobó que el efecto inhibitorio de la fusión mediado por quimoquinas se manifestaba solamente cuando el gen env usado se correspondía con aislamientos monocitotrópicos. Se pudo concluir, además, que la acción supresora de las quimoquinas era en realidad producto del bloqueo a la entrada viral. Por esa fecha pocos receptores de quimoquinas eran conocidos, el CCR5 se había clonado recientemente y se conocía que era receptor para las quimoquinas descritas por Gallo y Lusso [42]. Esto llevó a los investigadores a probar la capacidad del CCR5 de actuar como correceptor para la fusión en un ensayo similar al utilizado para la fusina. En este caso usaron células NIH 3T3 infectadas con virus vaccinias recombinantes para CD4/CCR5 y otras para CD4 solamente como control. Por otra parte usaron células HeLa con los genes lacZ y env del aislamiento monocitotrópico Ba-L. Así demostraron que la fusión era dependiente de la coexpresión en la superficie celular de CD4 y CCR5. Sin embargo, cuando midieron la capacidad de inhibición de la fusión por las diferentes quimoquinas encontraron que estas inhibían hasta un cierto límite. Esto deja posibilidad para especular sobre la existencia de otros correceptores. El mismo experimento se realizó sustituyendo el gen env. Se comprobó que CCR5 permitía la fusión para aquellas envolturas correspondientes a aislamientos monocitotrópicos o de comportamiento dual, pero no para los linfocitotrópicos. Además se realizaron experimentos de Northern blot y se halló la presencia de ARNm de CCR5 en macrófagos y células de sangre periféricas activadas con PHA, pero no en células que no son permisivas para aislamientos monocitotrópicos. El grupo de Landau ejecutó una serie de experimentos análogos [37]. Usando el gen de la luciferasa insertado en seudotipos virales (un mismo contexto viral con diferentes envolturas) se probó la capacidad de inhibión, de las quimoquinas descritas por Gallo, sobre la infección de la línea PM1. Esta línea es altamente susceptible de ser infectada por aislamientos tanto mono como linfocitotrópicos. Se comprobó que las quimoquinas sólo afectaban la infección cuando se trataba de envolturas de aislamientos monocitotrópicos. Se continuó con el clonaje de los receptores de quimoquinas conocidos, expresándolos en varias líneas humanas y murinas. Estas líneas así obtenidas se evaluaron en el mismo ensayo con los diferentes seudotipos virales. Las líneas que expresaron CD4 y los receptores: CCR1, CCR2b, CCR3 y CXCR4 fueron altamente resistentes a la infección por seudotipos para envolturas de aislamientos marcadamente monocitotrópicos. Sin embargo, las células que expresaron CCR5 fueron infectadas eficientemente por este tipo de aislamiento. En este caso la infección fue también estrictamente dependiente de la presencia de ambas moléculas: CCR5 y CD4. Estos resultados indicaron que CD4 y CCR5 actuaban concertadamente en la promoción de la entrada viral. Estos autores también hicieron estudios de Northen blot y llegaron a los mismos resultados que el grupo de Berger. Sin embargo, los receptores de quimoquinas CCR3 y CCR2b también han sido capaces de actuar como correceptores para un aislamiento primario de tropismo dual [39]. En particular el CCR3 puede actuar como correceptor para un grupo de aislamientos primarios [40]. Así pudiera ser posible que los virus linfocitotrópicos no evolucionen directamente a partir de aislamientos monocitotrópicos sino que exista un paso intermedio que se corresponde con virus de tropismo dual. Además, es predecible que toda terapia propensa a eliminar aislamientos virales con determinado tropismo tenderá por selección a promover la aparición de variantes de tropismo diferencial. A diferencia de la fusina no se ha descrito que CCR5 actúe por sí solo como receptor del virus y se ha mostrado que se requieren concentraciones elevadísimas de gp120 para promover su unión a células CCR5+, CD4- [43]. En el mismo estudio se utilizaron AcM contra varios sitios de la gp120 y se comprobó que varios de estos, incluyendo algunos contra V3, afectan la interacción con CCR5 sin afectar la unión a CD4. Esta es una evidencia en pro de la existencia de una región espacial, formada por varias zonas discretas de la envoltura, que es responsable de unirse al correceptor determinando el tropismo. Se ha comentado sobre la posible interacción correceptores/V3, argumentándose con una serie de observaciones [32]. Es conocido que en aislamientos de VIH-1 adaptados a líneas celulares los cambios en la región V3 provocan un cambio del uso de correceptor de CCR5 a CXCR4 [43, 44]. Se conoce que las mutaciones en el V3 que aumentan la carga neta positiva están asociadas con el cambio fenotípico de los aislamientos de VIH de no inductores (NSI) a inductores (SI) de sincitios [45, 46]. Así que se ha especulado que la carga neta negativa (-3) existente en el dominio extracelular tercero de CXCR4 podría interactuar electrostáticamente con la carga positiva del V3 [32]. Al ser la carga del dominio homólogo de CCR5 positiva (+2) se explicaría el comportamiento diferencial por los correceptores. Sin embargo, ni siquiera quienes han emitido estas ideas creen que la interacción a través de V3 sea la preponderante tomando en cuenta la poca similitud con el dominio análogo del VIH-2 [32]. Además hay que recordar un trabajo donde se muestra, para un aislamiento viral primario, que existen regiones independientes del V3 que condicionan el tropismo [47]. Lo más lógico es, nuevamente, que el tropismo esté determinado por una estructura espacial que involucra al V3, y además, otras regiones de la gp120. Se han publicado revisiones muy completas sobre los receptores de quimoquinas [31, 48]. Individuos CCR5- / CCR5- La mayoría de los aislamientos virales primarios de pacientes son monocitotrópicos no inductores de sincitios que no infectan líneas de linfocitos CD4+ [49]. Este fenotipo es sobre todo muy frecuente en los virus aislados poco tiempo después de la seroconversión [50-52]. Por todo esto se ha postulado que los aislamientos transmitidos por contacto sexual son fundamentalmente monocitotrópicos NSI [53]. Desde los primeros reportes del CCR5 como correceptor ya se esbozó la posibilidad de que personas de genotipos CCR5-/CCR5- fueran resistentes a la infección por vía natural del VIH-1 o al menos lentos progresores [41, 42]. Es conocido que dentro de los grupos de alto riesgo existen personas seronegativas cuyas células CD4 son resistentes a la infección por virus monocitotrópicos, no así para linfocitotrópicos [54]. Tan solo dos meses más tarde fue reportado, por dos grupos independientes, el hallazgo de un polimorfismo para la molécula CCR5 que confería resistencia a la infección por VIH-1 [55, 56]. Esta mutación en estado homocigótico se presenta en alrededor del 5% de los individuos resistentes a la infección. La variante descrita consta de una deleción de 32 pb (D32) codificante para el segundo dominio extracelular de la proteína. Esto provoca un corrimiento del marco de lectura que ocasiona la terminación temprana de la traducción. La proteína mutante consta de sólo 215 aa; los últimos 31 fuera del marco de lectura, a diferencia de la natural de 352 aa. Estudios encaminados a determinar la prevalencia de esta mutación en estado homocigótico en diferentes poblaciones humanas han mostrado reiteradamente que es más frecuente en caucasianos con alrededor del 1% [55, 57]. En africanos es aproximadamente diez veces menos frecuente [57]. Para el estado heterocigótico se ha hallado algo similar. En caucasianos se encuentra en, aproximadamente el 20%; en africanos e hispanos entre un 6 y 7%; en asiáticos en el 0,6%; en indios norteamericanos en el 12,6% y no se halló en indios suramericanos, ni en individuos africanos de la región occidental [58]. Otros alelos polimórficos han sido descritos para el CCR5 [57], sin embargo, ninguno de ellos ha sido correlacionado con protección. Diversos estudios han mostrado una alta correlación entre protección y el genotipo CCR5 D32 / CCR5 D32 [57-60]. Lo más importante es que se han estudiado más de 3 000 personas dentro de más de ocho cohortes. En todos estos trabajos se obtuvo que el 100% de los individuos D32 / D32 son seronegativos a pesar de pertenecer a grupos de alto riesgo. En estos estudios se encontró además que los individuos heterocigóticos (D32/+) tienden a progresar lentamente hacia la enfermedad una vez que contraen la infección, en comparación con individuos + / + [57-60]. Esto está en concordancia con los resultados obtenidos in vitro por Samson y cols. que mostraban que el estado heterocigótico de líneas celulares dificultaba su infección por aislamientos monocitotrópicos [55]. Sin embargo, la progresión lenta hacia la enfermedad de individuos heterocigóticos se ha reportado sólo para cohortes de homosexuales, no así para hemofílicos [57]. Esta observación puede ser explicada posiblemente por el hecho de que los hemofílicos estuvieron expuestos a altas dosis virales intravenosas antes que los pesquisajes de las donaciones fueran establecidos. También se han reportado resultados dispares en cuanto a diferencias estadísticas entre el número de heterocigóticos seropositivos y el de controles seronegativos. Samson y cols., reportaron una presencia significativamente menor de heterocigóticos dentro del grupo de seropositivos respecto del grupo control de seronegativos [55] lo cual sugiere un cierto nivel de protección contra la infección; sin embargo, otros autores no hallaron estas diferencias [57, 59, 61]. Sin embargo, parece que el número de correceptores en la superficie celular puede ser crítico para la infección. Se han reportado grandes diferencias en la expresión de CCR5 en la superficie celular entre los individuos +/+ [44] por lo que es altamente probable que el estado -/+ no implique, en algunos casos, un nivel de expresión significativamente diferente del que puede tener un homocigótico [62]. Es de notar el hecho que los aislamientos virales a partir de individuos heterocigóticos que progresan lentamente tienen tropismo por monocitos a diferencia de los aislamientos realizados, para el mismo grupo, en los que progresan más rápido que son linfocitotrópicos [60]. Esto evidencia que aún individuos D32/D32 no estarían protegidos de la infección por aislamientos linfocitotrópicos SI o aquellos de tropismo dual. Estudios in vitro han demostrado que la suposición era cierta [55, 63]. Posteriormente a los estudios realizados en cohortes, al menos tres publicaciones han reportado individuos D32/D32 seropositivos [64-66]. De los tres individuos reportados uno era hemofílico y recibió más de 500 000 unidades de factor VIII entre 1978 y 1984, tenía hepatitis B crónica y viremia de hepatitis C [64]; pero los otros dos son homosexuales sanos sin adicciones, ni historias de recepción de sangre por transfusiones [65, 66]. La caracterización de la secuencia del lazo V3 a través del tiempo mostró que el individuo hemofílico y otro de los individuos fueron infectados por aislamientos con tropismo linfocitario los cuales usan el CXCR4 como correceptor [65, 66]. En el caso del seropositivo restante no se han caracterizado los aislamientos que presenta. Tomando en cuenta los datos reportados queda claro que la ausencia de CCR5 no impide la infección por cepas linfocitotrópicas SI, ni por cepas capaces de usar correceptores alternativos como CCR3. La posibilidad de que existan mutaciones análogas para CXCR4 parece remota. El SDF-1 tiene como único receptor descrito a CXCR4. Los ratones transgénicos que no producen SDF-1 funcional mueren perinatalmente indicando el requerimiento absoluto de SDF-1 [67]. Por esto es lógico suponer que individuos CXCR4-/CXCR4- no sean viables. Patogénesis del VIH-1 Tomando en cuenta el reporte sobre la expresión diferencial de los correceptores en diferentes células del sistema inmune [68] Unutmaz y Littman han propuesto un modelo sencillo para explicar la patogénesis del VIH-1 [69]. Este modelo se basa en los siguientes hechos. Primero, CXCR4 se expresa predominantemente en células T vírgenes y CCR5 es expresado en células T activadas o de memoria [68]. Segundo, los linfocitos T vírgenes migran por órganos secundarios linfoides, mientras los activados y de memoria migran hacia órganos linfoides terciarios como pueden ser la piel y mucosas [70]. Tercero, que el VIH-1 es capaz de infectar macrófagos y células dendríticas [71], así como células T [72]. Según el modelo la infección natural transcurre por una serie de momentos (Figura 2). Probablemente, células dendríticas y macrófagos sean los primeros blancos de la infección en mucosa. La infección de las células presentadoras (APC) conllevaría a la consecuente activación de las células T de memoria con las que forman conjugados. Los linfocitos T activados tienen una expresión más alta de CCR5 que de CXCR4 [68]. Estas células son infectadas con mayor probabilidad por aislamientos que usen CCR5 como correceptor. Por esto es poco probable que los aislamientos linfocitotrópicos se diseminen al inicio de la infección. Tómese en cuenta además que las células activadas morirán, en su gran mayoría, en órganos linfoides terciarios sin diseminar más allá la infección. Sin embargo, las APC llevan la infección a órganos linfoides secundarios con su migración. Puesto que sólo los aislamientos monocitotrópicos son llevados a nódulos linfáticos eso explica por qué son los que se transmiten más fácilmente por vía natural. Como Bleul y cols. han mostrado, la expresión de los correceptores CXCR4 y CCR5 aumenta debido a la influencia de la interleuquina 2 (IL-2) [68]; la activación de las células T en nódulos linfáticos debería conllevar a su infección ahora por aislamientos dependientes de CXCR4 (linfocitotrópicos) que pueden surgir producto de mutaciones. Sin embargo, se postula que la alta expresión de SDF-1 por las células del estroma podría inhibir la entrada de los aislamientos linfocitotrópicos debido a la ocupación del correceptor [29, 30]. Sólo después de la destrucción de la arquitectura estromal sería posible que los aislamientos linfocitotrópicos infecten células T. Lo anterior explicaría por qué el predominio de aislamientos linfocitotrópicos ocurre en estados más avanzados de la infección [73]. Figura 2. Modelo propuesto por Unutmaz y Littman para explicar la patogénesis del VIH-1. A: entrada del virus a través de las mucosas; B: infección de células presentadoras a través de CCR5 y activación de linfocitos T de memoria; C: los linfocitos T son infectados pero muchos mueren después de su activación, las células presentadoras diseminan la infección a nódulos linfáticos; D: en ganglios muchos linfocitos T se infectan a través de CCR5 porque el SDF-1 producido por células del estroma mantiene ocupado el sitio de unión a CXCR4. La mólecula CCR5 aparece representada en la superficie de las células en color negro y CXCR4 en color blanco. Los correceptores CCR3 y CCR2b no han sido considerados para explicar la transmisión por vía natural del virus. El CCR3 se expresa en eosinófilos [74] y microglias [75]. Los primeros quedan descartados como blancos de la infección porque no expresan el marcador CD4. En el caso de las microglias, que sí expresan CD4, por su localización en el sistema nervioso central no se encuentran en la puerta de entrada de la infección. Sin embargo, después del paso del virus a la sangre, la infección de las microglias debe contribuir notablemente a las afecciones demenciales asociadas con el SIDA [76]. El caso de CCR2b que se expresa en monocitos sí pudiera ser relevante para la transmisión de algunas cepas [77]. Aparentemente la utilización de estos correceptores por algunas cepas parece ser producto de sus homologías con CCR5 y no debido a su uso diferencial respecto a éste. Es significativo que todas las cepas reportadas capaces de usar CCR3 y CCR2b como correceptores utilizan con alta eficiencia el CCR5. Algo realmente asombroso es que proteínas virales codificadas por otros virus pueden actuar como correceptores para el VIH-1. Así lo han demostrado Pleskoff y cols. [78]. Se conoce que varias proteínas homólogas a receptores de quimoquinas están codificadas en el genoma de herpesvirus [79-81]. Se han caracterizado en el genoma del citomegalovirus (CMV) los genes codificantes para US27, US28 y UL33 [82]. En particular US28 que puede actuar como receptor para las quimoquinas RANTES, MIP-1a y MIP-1b coincidiendo con CCR5 [26]. En el ya citado artículo se evidenció que US28 podía actuar como correceptor para aislamientos NSI y SI lo cual no se obtuvo para los casos de US27, ni UL33 [78]. Precisamente por ser el CMV común entre las personas infectadas por el VIH-1 [83] es, a la luz de estos experimentos, posible explicar al nivel molecular qué función podría desempeñar en la patogénesis del VIH. El CMV es capaz de infectar células CD4+ así como monocitos y macrófagos igualmente que el VIH-1 [84]. Tomando en cuenta todos estos datos sería perfectamente explicable la posible infección de individuos CCR5 D32/D32 por aislamientos primarios de fenotipo NSI previa infección con CMV. Resultados similares quizás se obtengan con el estudio de otros virus. Sirvan todas estas evidencias de excusa para aquellos que creyeron alguna vez que el VIH-1 no era el causante directo del SIDA sino un oportunista más [85]. Perspectivas futuras A pesar de la alta variabilidad en la secuencia de las proteínas de la envoltura del VIH-1 se ha podido establecer un sistema de clasificación en subtipos [86]. La mayoría de los aislamientos usados para el estudio de los correceptores se agrupan dentro del subtipo B predominante en EE UU y Europa. Sin embargo, Zhang y cols. han mostrado que lo mismo ocurre para aislamientos correspondientes a los otros subtipos. Ellos encontraron que la única variable que predice con gran acierto el uso diferencial de CCR5 o CXCR4 como correceptores es el fenotipo viral (SI o NSI) no así el subtipaje [87]. Esto deja claro que cualquier terapia basada en el uso de los correceptores tendrá aplicación global independientemente de los subtipos circulantes en cada región. A partir del trabajo publicado por el grupo de Gallo que mostraba que algunas quimoquinas eran capaces de inhibir la replicación viral [41], varios grupos encaminaron su trabajo a obtener quimoquinas antagonistas. Estas proteínas que carecen de actividad quimoatrayente pero no pierden la capacidad de unión a su ligando se han obtenido por modificación de las proteínas parentales [88, 89]. Se ha evidenciado para alguna de estas variantes que poseen una fuerza de unión a ligando mayor que la proteína natural, y que inhiben la entrada de aislamientos monocitotrópicos [89]. Hasta la actualidad no conocemos de ningún estudio clínico llevado a cabo con estas proteínas y sólo podemos prever su mejor efecto terapéutico en fases tempranas de la infección cuando predominan los aislamientos monocitotrópicos. Aunque no conocemos de la obtención de variantes modificadas similares para el SDF-1, ligando del CXCR4, creemos que la obtención de éstas y su uso dosificado podría constituirse en una terapia complementaria que limitaría en gran medida el predominio de los aislamientos linfotrópicos. El descubrimiento de los correceptores para el virus pudiera contribuir notablemente a la obtención de animales transgénicos capaces de simular eficientemente la enfermedad como ocurre en humanos. Entre los animales transgénicos para CD4 se han reportado ratones [90-92] y conejos [93, 94]; pero en ninguno el virus se replica a niveles importantes. Quizás la obtención del doble (CD4/CCR5 o CD4/CXCR4) o del triple transgénico (CD4/CCR5/CXCR4) conlleve a modelos adecuados. Se ha demostrado que líneas murinas expresando CD4 humano junto con la fusina de ratón son infectadas por aislamientos de VIH-1 linfocitotrópicos [95]. Sin embargo, se ha estudiado que en el caso murino las restricciones a que está sometido el virus no se aprecian solamente en la entrada [96]. Como en conejos no parece que existan esas otras restricciones es predecible que funcionará mejor como modelo [97]. Hay grupos en el mundo trabajando en esta dirección. La transferencia genética a células hematopoyéticas autólogas de móleculas codificantes de ARN sin sentido para afectar la traducción de los genes codificantes para los correceptores pudiera constituir una terapia para personas seropositivas al VIH-1. Sin embargo, esta tecnología se encuentra aún poco desarrollada. Los vectores retrovirales ensayados en simios y perros sólo brindan de un 0,1% a un 1% de eficiencia de transfección y lo mismo ha sucedido en ensayos clínicos con humanos [98]. Pero quizás por el gran efecto selectivo que ejercería la infección viral sobre el fenotipo de correceptores estas bajas frecuencias de transfección no impedirían el éxito de estas terapias. Sin embargo, sólo podemos prever como blanco de estas estrategias el CCR5 y no así el CXCR4 debido a que es el único receptor descrito para el SDF-1. Por esto el éxito de esta estrategia estaría condicionado a que no generasen quasiespecies capaces de utilizar CXCR4, CCR3 o CCR2b como correceptores. Se ha planteado la posibilidad de reconstituir el sistema inmune de individuos seropositivos sin SIDA a través del transplante de células de médula ósea proveniente de individuos D32/D32 [57]. El procedimiento habitual de transplante de médula conlleva la irradiación de los individuos previo a la inserción del tejido foráneo para evitar el rechazo. Esta práctica sería fatal si existiesen quasiespecies virales con tropismo linfocitario o dual en el individuo, puesto que las células implantadas serían infectadas, y por su bajo número sus funciones se verían afectadas en un corto período de tiempo. Quizás la combinación de las quimioterapias, tan efectivas, con alguna de estas estrategias sea un tratamiento mucho más eficiente que la aplicación exclusiva de algunas de éstas. Agradecimientos El autor quiere agradecer a sus compañeros del Departamento y en especial a la licenciada Mariela Quesada por suministrar parte de la bibliografía consultada y a la licenciada Ermita Reyes por su asistencia profesional. Además, a los doctores Eduardo Pentón y Carlos Duarte por revisar el manuscrito y aportar valiosas sugerencias. Referencias 1. Lane MC, Depper JL, Greene WC, Whalen G, Waldman T, Fauci AS. Qualitative analysis of immune function in patients with acquired immunodeficiency syndrome. N Engl J Med 1985;313:79-84. 2. McDougal JS, Kennedy MS, Sligh JM, Cort SP, Mawle A, Nicolson JKA. Binding of the human retrovirus HTLV-III/LAV to T4+ T cells by a complex of the 110K viral protein and the T4 molecule. Science 1986;231:382-5. 3. Tersmette M, DeGoede REY, Al BJM, Winkel IN, Gruters RA, Cuypers HT, et al. Differential syncytium-inducing capacity of human immunodeficiency virus isolates: frequent detection of syncytium-inducing isolates in patients with acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. J Virol 1988;62:2026-32. 4. Fenyö EM, Morrfeldt-Månson L, Chiodi F, Lind B, von Gegerfelt J, Albert E, et al. Distint replicative and cytopathic characteristics of human immunodeficiency virus isolates. J Virol 1988;62:4414-9. 5. Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 1986;233:215-9. 6. Gendelman HE, Baca L, Hysayni H, Turpin JA, Hoover DL, Meltzer MS. Macrophage-human immunodeficiency virus interaction: viral isolation and target cell tropism. AIDS 1990;4:221-8. 7. Maddon PJ, Dalgleish AG, McDougal JS, Clapham PR, Weiss RA, Axel R. The T4 gene encode the AIDS virus receptor and is expressed in the immune system and the brain. Cell 1986;47:333-48. 8. Dragic T, Picard L, Alizon M. Proteinase-resistant factors in human erythrocyte membranes mediated CD4-dependent fusion with cells expressing human immunodeficiency virus type 1 envelope glycoproteins. J Virol 1995;69:1013-8. 9. Dragic T, Charneau P, Clavel F, Alizon M. Complementation of murine cells for human immunodeficiency virus envelope/CD4-mediated fusion in human/murine heterokaryons. J Virol 1992;66:4794-802. 10. Broder CC, Dimitrov DS, Blumenthal R, Berger EA. The block of HIV-1 envelope glycoprotein-mediated membrane fusion in animal cells expresing human CD4 can be overcome by a human cell component(s). Virology 1993;193:483-91. 11. Fantini J, Cook DG, Nathanson N, Spitalnik SL, González-Scarano F. Infection of colonic epithelial cell lines by type 1 human immunodeficiency virus is associated with cell surface expression of galactosylceramide, a potencial alternative gp120 receptor. Proc Natl Acad Sci USA 1993;90:2700-4. 12. Harouse JH, Bhat S, Spitalnik SL, Laughlin M, Stefano K, Silberberg DH, et al. Inhibition of entry of HIV-1 in neural cell lines by antibodies against galactosyl ceramide. Science 1991;253:320-3. 13. Homsy J, Meyer M, Tateno M, Clarkson S, Levy J. The Fc and not the CD4 receptor mediates antibody enhancement of HIV infection in human cells. Science 1989;244:1357-60. 14. Hildreth JE, Orentas RJ. Involvement of a leukocyte adhesion receptor (LFA-1) in HIV-induced syncytium formation. Science 1989;244:1075-8. 15. Avril LE, DiMartino-Ferrer M, Barin F, Gauthier F. Interaction between a membrane-associated serine proteinase of U-937 monocytes and peptides from the V3 loop of the human immunodeficiency virus type 1 (HIV-1) gp120 envelope glycoprotein. FEBS Lett 1993;317:167-72. 16. Callebaut C, Krust B, Jacotot E, Hovanessian AG. T-cell activation antigen, CD26, as a cofactor for entry of HIV in CD4+ cells. Science 1993;262:2045-50. 17. Sato AI, Balamuth FB, Ugen KE, Williams WV, Weiner DB. Identification of CD7 glycoprotein as an accesory molecule in HIV-1 mediated syncytium formation and cell-free infection. J Immunol 1994;152:5142-52. 18. Dukes CS, Yu Y, Rivadeneira ED, Sauls DL, Laio HX, Haynes BF, et al. Cellular CD44S as a determinant of human immunodeficiency virus type 1 infection and cellular tropism. J Virol 1995;69:4000-5. 19. Sozzani S, Locati M, Allavena P, Van Damme J, Mantovani A. Chemokines: a sperfamily of chemotactic cytokines. Int J Clin Lab Res 1996;26:69-82. 20. Power CA, Wells TNC. Cloning and characterization of human chemokine receptors. TiPS 1996;17:209-12. 21. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996;272:872-7. 22. Baiocchi M, Olivetta E, Chelucci C, Santarcangelo AC, Bona R, d´Aloja P, et al. Human immunodeficiency virus (HIV)-resistant CD4+ UT-7 megakaryocytic human cell line becomes highly HIV-1 and HIV-2 susceptible upon CXCR4 transfection: induction of cell differentiation by HIV-1 infection. Blood 1997;89:2670-8. 23. Herzog H, Hort YJ, Shine J, Selbie LA. Molecular cloning, characterization, and localization of the human homolog to the reported bovine NPY Y3 receptor: lack of NYP binding and activation. DNA Cell Biol 1993;12:465-71. 24. Jazin EE, Yoo H, Bloqvist AG, Yee F, Weng G, Walker MW, et al. A proposed bovine neuropeptide Y (NPY) receptor cDNA clone, or its human homologue, confers neither NPY binding sites nor NPY responsiveness on transfected cells. Regul Pept 1993;47:247-58. 25. Federsppiel B, Melhado IG, Duncan AMV, Delaney A, Schappert K, Clark-Lewis I, et al. Molecular cloning of the cDNA and chromosomal localization of the gene for a putative seven-transmembrane segment (7-TMS) receptor isolated from human spleen. Genomics 1993;16:707-12. 26. Neote K, DiGregorio D, Mak JY, Horuk R, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell 1993;72:415-25. 27. Nomura H, Nielsen BW, Matsushima K. Molecular cloning of cDNAs encoding a LD78 receptor and putative leukocyte chemotactic peptide receptors. Int Immunol 1993;5:1239-49. 28. Letscher M, Geiser T, O´Relly T, Zwahlen R, Baggiolini M, Moser B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J Biol Chem 1994;289:232. 29. Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Seisdedos FA, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 1996;382:833-5. 30. Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996;382:829-33. 31. Doranz BJ, Berson JF, Rucker J, Doms RW. Chemokine receptors as fusion cofactors for human immunodeficiency virus type 1 (HIV-1). Immunol Res 1997;16:15-28. 32. Brelot A, Heveker N, Pleskoff O, Sol N, Alizon M. Role of the first and third extracellular domains of CXCR-4 in human immunodeficiency virus co-receptor activity. J Virol 1997;71:4744-51. 33. Pleskoff O, Sol N, Labrosse B, Alizon M. Human immunodeficiency virus strains differ in their ability to infect CD4+ cells expressing the rat homolog of CXCR-4 (Fusin). J Virol 1997;71:3259-62. 34. Hesselgesser J, Halks-Miller M, DelVecchio V, Peiper SC, Hoxie J, Kolson DL, et al. CD4-independent association between HIV-1 gp120 and CXCR4: functional chemokine receptors are expressed in humans neurons. Curr Biol 1997;7:112-21. 35. Endres MJ, Clapham PR, Marsh M, Ahuja M, Turner JD, McKnigth A, et al. CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell 1996;87:745-56. 36. Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, et al. CC CKR-5: a RANTES, MIP-1a, MIP-1b receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996;272:1955-8. 37. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996;381:661-6. 38. Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996;381:667-73. 39. Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, et al. A dual-tropic primary HIV-1 isolate that uses fusin and the b-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 1996;85:1149-58. 40. Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, et al. The b-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 1996;85:1135-48. 41. Cocchi F, DeVico AL, Demo AG, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1a, and MIP-1b as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995;270:1811-5. 42. Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry 1996;35:3362-7. 43. Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, et al. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 1996; 384:184-7. 44. Speck RF, Wehrly K, Platt EJ, Atchison RE, Charo IF, Kabat D, et al. Selective employment of chemokine receptors as human immunodeficiency virus type 1 co-receptors determined by individual amino acids within the envelope V3 loop. J of Virol 1997;71:7136-9. 45. Callahan L. HIV-1 virion-cell interactions: an electrostatic model of pathogenicity and syncytium formation. AIDS Res Hum Retroviruses 1994;10:231-3. 46. Fouchier RAM, Groenink M, Kootstra NA, Tersmette M, Huisman HG, Miedema F, et al. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 pg120 molecule. J Virol 1992;66:3183-3187. 47. Kim F, Kolson DL, Balliet JW, Srinavasan A, Collman RG. V3-independent determinants of macrophage tropism in a primary human immunodeficiency virus type 1 isolate. J of Virol 1995;69:1755-61. 48. Broder CC, Collman RG. Chemokine receptors and HIV. J Leukoc Biol1997;62:20-9. 49. Cheng-Mayer C, Seto D, Tateno M, Levy JA. Biological features of HIV that correlate with virulence in the host. Science 1988;240:80-2. 50. Conner RI, Ho DD. Human immunodeficiency virus type 1 variants with increased replicative capacity develop during the asyntomatic stage before disease progression. J Virol 1994;68:4400-8. 51. Conner RI, Mohri H, Cao Y, Ho DD. Increased viral burden and cytopathicity correlate temporally with CD4+ T lymphocyte decline and clinical progression in human immunodeficiency virus type 1-infected individuals. J Virol 1993;67:1772-7. 52. Roos MTL, Lange JMA, de Goede REY, Coutinho RA, Schellekens PTA, Miedema F, et al. Viral phenotype and immune response in primary human immunodeficiency virus type 1 infection. J Infect Dis 1992;165:427-32. 53. Zhu T, Wang H, Nam N, Cao DS, Koup RA, Ho DD. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 1993;261:1179-81. 54. Paxton WA, Martin SR, Tse D, O´Brien TR, Skurnick J, Van-Devanter NL, et al. Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposure. Nature Med 1996;2:412-7. 55. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996;382:722-5. 56. Rong L, Paxton WA, Choe S, Ceradni D, Martin SR, Horuk R, et al. Homozygous defect in HIV-1 co-receptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996;86:367-77. 57. Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 1996;273:1856-62. 58. Zimmerman PA, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, Combadiere C, et al. Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk. Mol Med 1997;3:23-36. 59. Olsen JE, Iversen AKN, Garred P, Koppelhus U, Pedersen C, Benfield TL, et al. Heterozygosity for a deletion in the CKR-5 gene leads to prolonged AIDS-free survival and slower CD4 T-cell decline in a cohort of HIV-seropositive individuals. AIDS 1997;11:305-10. 60. Michael NL, Chang G, Louie LG, Mascola JR, Dondero D, Birx DL, et al. The role of viral phenotype and CCR-5 gene defects in HIV-1 transmission and disease progression. Nature Med 1997;3:338-40. 61. Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T, et al. The role of a mutant CCR5 allele in the HIV-1 transmission and disease progression. Nature Med 1996;2:1240-3. 62. Moore JP. Co-receptors: implications for HIV pathogenesis and therapy. Science 1997;276:51-2. 63. Rana S, Besson G, Cook DG, Rucker J, Smyth RJ, Yi Y, et al. Role of CCR5 in infection of primary macrophages and lymphocytes by macrophage-tropic strains of human immunodeficiency virus: resistance to patient-derived and prototype isolates resulting from the Dccr5 mutation. J of Virol 1997;71:3219-27. 64. Biti R, Ffrench R, Young J, Bennetts B, Stewart G. HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nature Med 1997;3:252-3. 65. O´Brien TR, Winkler C, Dean M, Nelson JAE, Carrington M, Michael NL, et al. HIV-1 infection in a man homozygous for CCR5D32. The Lancet 1997;349:1219. 66. Theodorou I, Meyer L, Magierowska M, Katlama C, Rouzioux C, and the Seroco Study Group. HIV-1 infection in an individual homozygous for CCR5D32. The Lancet 1997;349:1219-20. 67. Nagasaw T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996;382:635-8. 68. Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV co-receptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci USA 1997;94:1925-30. 69. Unutmaz D, Littman DR. Expression pattern of HIV-1 co-receptors on T cells: implications for viral transmisssion and lymphocyte homing. Proc Natl Acad Sci USA 1997;94:1615-8. 70. Ford WL. Lymphocyte migration and immune responses. Prog Allergy 1975;19:1-59. 71. Cameron PU, Freudenthal PS, Barker JM, Gezelter S, Inaba K, Steinman RM. Dendritic cells exposed to human immunodeficiency virus type 1 transmit a vigorous cytopathic infection to CD4+ T cells. Science 1992;257:383-7. 72. Ramilo O, Bell KD, Uhr JW, Vitetta ES. Role of CD25+ and CD25- T cells in acute HIV infection in vitro. J of Immunol 1993;150:5202-8. 73. Connor RI, Ho DD. Human immunodeficiency virus type 1 variants with increased replicative capacity develop during the asymptomatic stage before disease progression. J Virol 1994;68:4400-8. 74. Combadiere C, Ahuja SK, Murphy PM. Cloning and functional expression of a human eosinophil CC chemokine receptor. J Biol Chem 1995;270:16491-4. 75. He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 1997;385:645-9. 76. Price RW. The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science 1988;239:586-91. 77. Combadiere C, Ahuja SK, Damme JV, Tiffany HL, Gao JL, Murphy PM. Monocyte chemoattractant protein-3 is a functional ligand for CC chemokine receptors 1 and 2B. J Biol Chem 1995;270:29671-5. 78. Pleskoff O, Tréboute C, Brelot A, Heveker N, Seman M, Alizon M. Identification of a chemokine receptor encoded by human cytomegalovirus as a cofactor for HIV-1 entry. Science 1997;276:1874-8. 79. Ahuja SK, Murphy PM. Molecular piracy of mammalian interleukin-8 receptor type B by Herpesvirus Saimiri. J Biol Chem 1993; 268:20691-4. 80. Nicholas J. Determination and analysis of the complete nucleotide sequence of human Herpesvirus 7. J Virol 1996;70:5975-89. 81. Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 1997;385:347-50. 82. Chee MS, Satchwell SC, Preddie E, Weston KM, Barrell BG. Human cytomegalovirus encodes three G protein-coupled receptor homologues. Nature 1990;344:774-7. 83. Danner SA. Management of cytomegalovirus disease. AIDS 1995;9(suppl 2):S3-S8. 84. Rice GPA, Schrier RD, Oldstone MBA. Cytomegalovirus infects human lymphocytes and monocytes: virus expression is restricted to immediate-early gene products. Proc Natl Acad Sci USA 1984;81:6134-8. 85. Duesberg P. HIV and AIDS. Science 1993; 260:1705. 86. Myers G, Korber BTM, Smith RF, Wain-Hobson S & Pavlakis GN (eds): Human Retroviruses and AIDS 1993. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, New Mexico, 1993. 87. Zhang L, Huang Y, He T, Cao Y, Ho D. HIV-1 subtype and second-receptor use. Nature 1996;383:768. 88. Arenzana-Seisdedos F, Virelizier JL, Rousset D, Clark-Lewis I, Loestscher P, Moser B, et al. HIV blocked by HIV chemokine antagonist. Nature 1996;283:400. 89. Simmons G, Clapham PR, Picard L, Offord RE, Rosenkilde MM, Schwartz TW, et al. Potent inhibition of HIV-1 infectivity in macrofages and lymphocytes by a novel CCR5 antagonist. Science 1997;276:276-9. 90. Gillespie FP, Doros L, Vitale J, Blackwell C, Gosselin J, Snyder BW, et al. Tissue-specific expression of human CD4 in transgenic mice. Mol Cell Biol 1993;13:2952-8. 91. Killeen N, Sawada S, Littman DR. Regulated expression of human CD4 rescue helper T-cell development in mice lacking expression of endogenous CD4. Eur Mol Biol Org 1993;12:1547-53. 92. Hanna Z, Simard C, Laperriere A, Jolicoeur P. Specific expression of the human CD4 gene in mature CD4(+) CD8 (-) an immature CD4(+) CD8(+) T cells and in macrophages of transgenic mice. Mol Cell Biol 1994;14:1084-94. 93. Snyder BW, Vitale J, Milos P, Gosselin J, Gillespie F, Ebert K, et al. Developmental and tissue-specific expression of human CD4 in transgenic rabbits. Mol Reprod Dev 1995; 40:419-28. 94. Dunn CS, Mehtali M, Houdebine LM, Gut JP, Kirn A, Aubertin AM. Human immunodeficiency virus type 1 infection of human CD4-transgenic rabbits. J Gen Virol 1995;76:1327-36. 95. Tachibana K, Nakajima T, Sato A, Igarashi K, Shida H, Iizasa H, et al. CXCR4/fusin is not species-specific barrier in murine cells for HIV-1 entry. J Exp Med 1997;185:1865-70. 96. Trono D. Molecular biology of HIV. Clin Lab Med 1994;14:203-20. 97. Cho S, Kindt TJ, Zhao TM, Sawasdikosol S, Hague BF. Replication of HIV type 1 in rabbit cell lines is not limited by deficiencies in tat, rev, or long terminal repeat function. AIDS Res Hum Retroviruses 1995;11:1487-93. 98. Kohn DB. Gene therapy for haematopoietic and lymphoid disorders. Clin Exp Immunol 1997;107 Suppl 1:54-7. Copyright 1998 Elfos Scientiae The following images related to this document are available:Line drawing images[ba98035a.gif] [ba98035b.gif] |
| |||||||||

{kind=link}
{kind=link}