 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
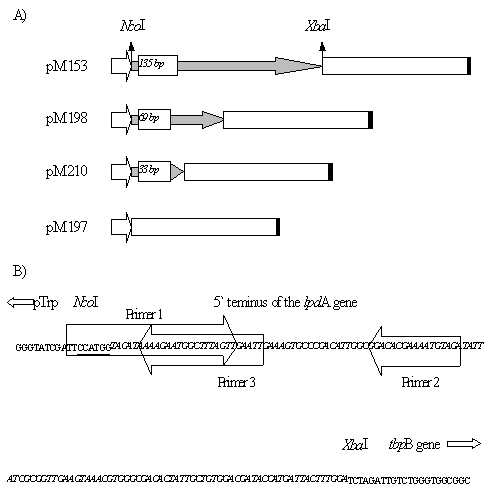
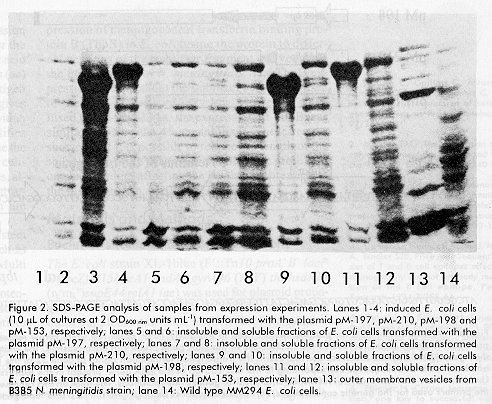
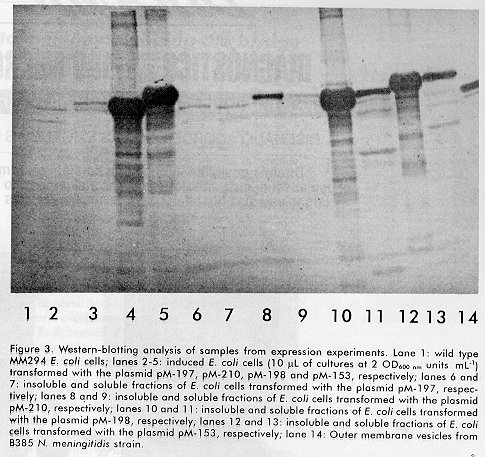
Biotecnología Aplicada 1998;15:254-257 INFLUENCE OF THE LENGTH OF THE P64k PROTEIN N-TERMINUS STABILIZING PEPTIDE ON THE EXPRESSION IN Escherichia coli OF THE FUSED TbpB FROM Neisseria meningitidis@ Tamara Menéndez, Luis M Alonso, Mairet Pérez, Rolando Pajón, Ricardo SilvaCentro de Ingeniería Genética y Biotecnología. Ave 31 entre 158 y 190. AP 6162, CP 10600, Cubanacán, Habana, Cuba. E-mail: Ricardo.Silva@cigb.edu.cu
Code Number: BA98042
ABSTRACT In order to improve the previously patented system for the expression of heterologous antigens in Escherichia coli, the possibility of reducing the length of the 45 amino acid (aa) stabilizer peptide from the P64k protein of Neisseria meningitidis, was investigated. The TbpB protein from N. meningitidis was expressed fused to the first 23 and 11 aa from the P64k protein and without the stabilizer peptide. The levels of expression of the TbpB protein were very low, only detectable by Western blotting when expressed without the stabilizer peptide or fused to the first 11 aa from the P64k protein. High levels of expression were achieved when the protein was expressed fused to the first 23 aa from the P64k protein. These results showed that it is possible to reduce the length of the stabilizer peptide derived from the P64k protein to one half without affecting the levels of expression of the fusion protein. Keywords: P64k protein, transferrin binding proteins, TbpB, expression system, fusion proteins RESUMEN Con el objetivo de optimizar el sistema de expresión de antígenos heterólogos en Escherichia coli, patentado previamente, se investigó la posibilidad de reducir la talla del fragmento estabilizador de 45 aminoácidos (aa) provenientes de la proteína P64k de Neisseria meningitidis. La proteína TbpB de N. meningitidis se expresó en E.coli fusionada a los primeros 23 y 11 aa de la proteína P64k y sin el fragmento estabilizador. En los dos últimos casos se obtuvieron niveles bajos de expresión de TbpB, solo detectables mediante Western blotting. Se obtuvieron altos niveles de expresión de TbpB, cuando se expresó fusionada a los primeros 23 aa de la proteína P64k. Estos resultados muestran que es posible reducir hasta la mitad la talla del fragmento estabilizador derivado de la proteína P64k sin afectar los niveles de expresión de la proteína de fusión. Palabras claves: proteína P64k, proteínas de unión a transferrina, TbpB, sistemas de expresión, proteínas de fusión Introduction In a previously developed Escherichia coli expression system [1], the nucleotide sequence encoding for the specific protein is cloned in a frame with a nucleic acid sequence that codifies for the first 45 amino acids (aa) derived from the amino terminus of the P64k antigen from the Neisseria meningitidis strain B385. This gives rise to high levels of expression of a fusion protein within the cytoplasm of the cell. The method also simplifies and cheapens the initial purification steps, because the protein is obtained with relatively high purity and concentration due to the formation of inclusion bodies, although in these cases different methods for the subsequent renaturation of the recombinant protein are needed [1-3]. We have successfully used our expression system for the production of heterologous proteins, such as class 1 and OpC proteins of N. meningitidis and Multi epitopic peptides derived from HIV-1 [1]. However, when the heterologous proteins are potential vaccine candidates, as in our case, there are some disadvantages concerning the use of stabilizer peptides that constitute a part of the fusion protein [4-6]. Therefore, we were interested in investigating the possibility of modifying our expression system in order to reduce the length of the fusion stabilizing peptide. In this study we have characterized the influence of the stabilizing peptide length on the levels of expression of meningococcal transferrin binding protein B (TbpB) in E.coli fusing the protein to different versions of the stabilizer fragment derived from the P64k protein of N.meningitidis. The TbpB expression levels without the stabilizer peptide or fused to the first 11 aa were very low. However, when fused to the first 23 aa, the expression of TbpB was similar to using the 45 aa stabilizer. These results showed that it is possible to reduce the length of the stabilizer peptide derived from the P64k protein to one half without affecting the levels of expression of the fusion protein. Materials and Methods Bacterial strains and growth conditions The E.coli strain XL-1blue (F´::Tn10 proA+B+ lacIq D(lacZ)M15/recA1 endA1 gyrA96 (NaIr) thi hsdR17 (rk-mk+)supE44 relA1 lac) was used for plasmid propagation and grown overnight at 37°C in an LB medium supplemented with ampicillin (50 mg/mL-1) when carried plasmid. The E.coli strain MM294 (F- endA1 hsdR17(rk-mk+)supE44 thi-1 relA1? rfbD1? spoT1?), was used for expression analysis and grown overnight at 37°C in an LB medium supplemented with ampicillin (50mg/mL-1) or kanamycin (50mg/mL-1), when required. Genetic constructions Figure 1A shows the features of the plasmids used in the present work. pM-153: It contains the tbpB gene from the N.meninigitidis strain B385 fused to the first 135 bp of the lpdA gene from N.meningitidis under the control of the tryptophan promoter (ptrp) from E.coli. The vector also includes the transcription terminator of gene 32 of bacteriophage T4 and the resistance genes for ampicillin and kanamycin as selection markers. pM-197: The expression plasmid pM-153 was restricted NcoI-XbaI, the ends filled using the Klenow fragment of DNA Polymerase I and ligated using T4 DNA ligase. pM-198 and pM-210: The Polymerase chain reaction (PCR), for the amplification of the first 69 base pairs (bp) of the lpdA gene from the N.meningitidis strain B385, was performed under the following conditions: 30 cycles of 95ºC for 1 min, 58ºC for 1 min and 72ºC for 30 sec. The reaction components were: 1mg of chromosomal DNA from the strain B385; 50 pmol of primers 1, (5´...TTCCATGGTAGATA AAAGAATGGCTTT AGTT...3´) and 2 (5´...CATCTA GATCTACATTT TCGTGTCC...3´); 200 mm of each deoxyribonucleoside triphosphate (dNTPs); PCR buffer (10 mM KCL; 20mM Tris-HCl, pH 8.8; 10mM (NH4)2SO4; 2mM MgSO4; 0.1% Triton X-100), double distilled water to a final volume of 75 mL and 1Unit per reaction of Thermusaquaticus DNA Polymerase. The fragment corresponding to the first 33 bp of the lpdA gene was obtained by hybridization of primers 1 and 3 (5´...CATCTAG AAATTCAACTAAAGCCATTCTTTT...3´) and further extension with T.aquaticus DNA Polymerase 1h at 72ºC, in the above described conditions. Both fragments were digested with NcoI and XbaI, before ligation into the pM-153 vector, previously digested with the same enzymes. Figure 1B shows the sites of hybridization in the lpdA gene sequence of the primers used for the construction of pM198 and pM210. The restriction enzymes and the T4 DNA ligase were purchased from New England Biolabs (MA, USA) and used according to the manufacturer`s recommendations. The synthetic primers and the T.aquaticus DNA polymerase were purchased from Heber Biotec (Cuba). The confirmatory DNA sequences for the constructs pM197, pM198 and pM210 were done following the Sanger´s method [7] using the Sequenase 2.0 kit (USB, USA). Standard methods were used for plasmid and chromosomal DNA preparation, restriction endonuclease analysis and ligations, and were carried out as described [8]. Ultrasonic cell disruption For disruption, E.coli cells were suspended at a 1:10 (w:v) ratio in TE buffer (10 mM Tris-HCl and 1 mM EDTA pH 8) and subjected to 5 cycles of ultrasonic disruption of 1 min each. After the disruption, the cellular debris was collected by centrifugation. The pellets and supernatants were stored separately at 4°C for further analysis. The process was carried out at 4°C. Protein concentration was determined following the Lowry method [9] and 20 mg of protein from both samples were analyzed by SDS-PAGE and Western blotting. SDS-PAGE and Western blotting The OD600nm was measured and the cell pellets of 1mL of each sample were suspended in Laemmli sample buffer (0.06 M Tris-HCl, pH 6.9; 5% (v/v) b-mercaptoethanol; 4% (w/v) Sodium Dodecyl Sulphate (SDS); 10% (v/v) Glycerol) to a concentration of 2 OD units mL-1. Ten mL of each sample were loaded per lane and subjected to 10% SDS-PAGE as described [10]. Proteins were visualized with Coomassie Brilliant Blue staining or, for immunoblotting, they were transferred from acrylamide gels to 0.45 mm pore size nitrocellulose membranes. The membranes were blocked with 5% (w/v) skimmed milk in PBS (0.14 M NaCl, 0.003 M KCl, 0.008 M Na2HPO4 and 0.0015 M KH2PO4) containing 0.05% (v/v) Tween 20 (blocking solution), washed once with PBS/0.05% Tween 20 and incubated 1 h at 37°C with a blocking solution containing an immune rabbit serum directed against the TbpA/TbpB complex of N.meningitidis strain M982, kindly provided by Dr Quentin-Millet (Pasteur Merieux Connaught, France). After three washes with PBS/0.05% Tween 20, membranes were incubated for 1 h at 37°C with Donkey IgGs anti rabbit immunoglobulins conjugated with Horseradish peroxidase (HRPO) (Amersham, England). The membranes were washed again and the reactions were developed with 50 mM of NaAc buffer pH 5.2, 0.2 mg/mL of 3-amino-9-ethyl-carbazole and hydrogen peroxide 0.015% (v/v) for 5 min. Results and Discussion In a previous study [1], we developed an E.coli expression system where heterologous proteins were expressed fused to the first 45 aa of the P64k protein from N.meningitidis. The use of this expression system is suitable because the levels of expression of the heterologous proteins fused to the fragment derived from the P64k protein are comparable to those achieved when the same proteins are expressed fused to stabilizing fragments derived from the human Interleukine-2 (IL-2) (1). Furthermore, the use of monoclonal antibodies (MAbs) that recognize the stabilizing fragment from the P64k protein, enables the detection of a great variety of fusion proteins without the previous availability of immunologic probes against each polypeptide to be expressed. The MAbs also permits the affinity purification of the expressed protein if it is immobilised in a chromatographic resin [1]. The system is also advantageous when the proteins to be expressed are vaccine candidates. The stabilizer fragment derived from the P64k protein is poorly immunogenic and hence exerts a minim influence on the immunogenicity of the polypeptides to which it is fused. Finally, no mammalian protein with meaningful similarities with the stabilizer peptide derived from the P64k protein has been identified [1]. However, the use of stabilizer peptides that form part of the fusion protein has certain disadvantages if the latter is a vaccine candidate. The presence of foreign sequences can alter the native order of B and T cell epitopes [4] or their processing and presentation by the antigen presenting cells [5], or even affect the immunogenicity of the candidate by the epitope- specific suppression phenomenon [6]. In some cases, small fragments that still stabilize the expression have been defined. For example, the use, as stabilizer peptides, of the first 95, 58 and 38 aa derived from the human IL-2 has been patented [11-13]. In the present paper we investigated the possibility of improving our expression system by reducing the length of the fusion partner. We evaluated the TbpB protein, as a model in our studies. TbpB from N. meningitidis is an attractive vaccine candidate [reviewed in 14]. For us, the best choice would be to produce the full-length recombinant TbpB protein in E.coli, without any fusion partner, as a soluble protein and with high levels of protein expression. But usually when heterologous proteins are expressed in high levels, they are produced in an insoluble form as inclusion bodies [15]. Therefore it is advisable to define small fragments that still stabilize the protein expression and could reduce the detrimental effects described above. In previous experiments we expressed the tbpB gene from the N.meningitidis strain B385 fused to the first 45 aa of P64k protein of N.meningitidis. The expression plasmid used was the pM-153 shown in Figure 1. The levels of expression achieved accounted for 30% of the total cellular proteins (Figure 2, lane 4). These levels of expression are suitable for scaling up the purification process of the recombinant protein. In this case the protein was mainly detected in the insoluble fraction of the cell (Figure 2, lane 11 and Figure 3, lane 12). Figure 1. A: expression units for plasmid vectors used in this study. In all cases the white arrow represents the tryptophan promoter, the gray arrow represents the nucleotides of the lpdA gene present in each plasmid, the white box represents the tbpB gene (2088 bp) and the black box represents the transcription termination signal of gene 32 of the T4 phage. B: in italics and bold is the DNA sequence corresponding to the first 135 bp of the lpdA gene present in the expression plasmid pM153. Underlined are the NcoI and XbaI restriction sites used for genetic manipulations. The thick arrows represent the localization of the primers used for the genetic constructions. Part of the sequences belonging to the ptrp and tbpB genes are also shown. Figure 2. SDS-PAGE analysis of samples from expression experiments. Lanes 1-4: induced E.coli cells (10 mL of cultures at 2 OD600 nm units mL-1) transformed with the plasmid pM-197, pM-210, pM-198 and pM-153, respectively; lanes 5 and 6: insoluble and soluble fractions of E. coli cells transformed with the plasmid pM-197, respectively; lanes 7 and 8: insoluble and soluble fractions of E. coli cells transformed with the plasmid pM-210, respectively; lanes 9 and 10: insoluble and soluble fractions of E. coli cells transformed with the plasmid pM-198, respectively; lanes 11 and 12: insoluble and soluble fractions of E. coli cells transformed with the plasmid pM-153, respectively; lane 13: outer membrane vesicles from B385 N. meningitidis strain; lane 14: Wild type MM294 E.coli cells. The importance of the use of the stabilizer sequence was confirmed, because the tbpB gene cloned directly after the ptrp (plasmid pM-197 in Figure1) was expressed at levels only detectable by Western-blotting (Figure 2, lane 1 and Figure 3, lane 2). The protein was detected mainly in the soluble fraction of the disrupted cells (Figure 3, lanes 6 and 7). Figure 3. Western-blotting analysis of samples from expression experiments. Lane 1: wild type MM294 E. coli cells; lanes 2-5: induced E. coli cells (10 mL of cultures at 2 OD600 nm unitsmL-1) transformed with the plasmid pM-197, pM-210, pM-198 and pM-153, respectively; lanes 6 and 7: insoluble and soluble fractions of E. coli cells transformed with the plasmid pM-197, respectively; lanes 8 and 9: insoluble and soluble fractions of E. coli cells transformed with the plasmid pM-210, respectively; lanes 10 and 11: insoluble and soluble fractions of E. coli cells transformed with the plasmid pM-198, respectively; lanes 12 and 13: insoluble and soluble fractions of E. coli cells transformed with the plasmid pM-153, respectively; lane 14: Outer membrane vesicles from B385 N. meningitidis strain. To try to reduce the length of the stabilizer fragment to an extent still capable of stabilizing the expression, the tbpB gene was expressed fused to the first 23 aa from the P64k protein. The E.coli cells transformed with the plasmid pM-198 (Figure 1) showed levels of expression of the fusion protein similar to those obtained when the TbpB was expressed fused to the first 45 aa from the P64k protein (Figure 2, lanes 3 and 4 and Figure 3, lanes 4 and 5). In this case the major part of the fusion protein was present, as in the case of the TbpB with the full-length stabilizer, in the insoluble fraction of the cell (Figures 2, lane 9 and Figure 3, lane 10). To reduce, even more, the length of the stabilizer peptide, the tbpB gene was expressed fused to the first 11 aa from the P64k protein. The E.coli cells transformed with the corresponding expression plasmid (pM-210 in Figure 1) showed levels of expression of the fusion protein only detectable by Western-blotting (Figure 2, lane 2 and Figure 3, lane 3). Ultrasonic disruption and further analysis of cellular fractions showed that the fusion protein was present mainly in the soluble fraction of the cells (Figure 3, lanes 8 and 9). These results were similar to those obtained when the TbpB protein was expressed without the stabilizer fragment. Certain conclusions can be drawn from this study: 1. The stabilizer fragment derived from the P64k protein is important to increase the heterologous protein expression. 2. It is possible to reduce the length of the original stabilizer fragment from 45 to 23 aa without affecting the levels of expression of the recombinant protein. The fragment of 11 aa is not enough to stabilize the protein expression. The reduction of the length of the stabilizer peptide is important in the case of the expression of vaccine candidates where the recombinant protein must be as similar as possible to the natural protein. 3. Besides the enhancing effect on expression levels, the N-terminus fragment of the P64k protein might have some influence in determining the solubility status of the specific fusion protein within the bacterium. 4. With the modification of the expression system developed here it is possible to produce recombinant proteins in E.coli with yields compatible with the scaling up of recombinant products. Acknowledgments The authors want to thank Dr. Gerardo Guillén, Dr. Alexis Mussachio and Dr. Conzuelo Nazábal for the critical reading of the manuscript. References 1. Duarte C, Guillén G, Álvarez A, Carpio E, Quintana D, Gómez C, et al. System for the expression of heterologous antigens as fusion proteins. WO 97/26359. 2. Marston FAO. The purification of eucaryotic polypeptides synthesized in Escherichia coli. Biochem J 1986;240:1-12. 3. Fischer B, Sumner I, Goodenough P. Isolation, renaturation and formation of disulfide bonds of eukaryotic proteins expressed in Escherichia coli as inclusion bodies. Biotechnol Bioeng 1993;41:3-13. 4. Denton G, Hudecz F, Kajtár J, Murray A, Tendler SJB, Price MR. Sequential order of T and B cell epitopes affects immunogenicity but not antibody recognition of the B cell epitope. Peptide Research 1994;7:258-64. 5. Del Val M, Schlicht H, Ruppert T, Reddehase MJ, Koszinowski UH. Efficient processing of an antigenic sequence for presentation by MHC class1 molecules depends on its neighboring residues in the protein. Cell 1991;66:1145-53. 6. Etlinger H. Carrier sequence selection, one key to successful vaccine. Immunol Today 1992;13:52-5. 7. Sanger F, Niclen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 1977;74:5463-5467. 8. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual 1989. 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 9. Lowry OH, Rosemmbrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265-275. 10. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227: 680-685. 11. Habermann P. Hirudin derivatives. Hoechst Aktiengesellschaft. European Patent Application No 0468539. 12. Novoa LI, Machado JA, Fernández JR, Benítez JV, Narciandi RE, Rodríguez JL etal. Method for the expression of heterologous proteins produced in fused form in E.coli, use thereof, expression vectors and recombinant strains. Centro de Ingeniería Genética y Biotecnología. European Patent Application No. 0416673. 13. Habermann P. Fusion proteins with an eukariotic ballast sequence. Hoechst Aktiengesellschaft. European Patent Application No. 0229998 14. Ala´Aldeen DAA. Transferrin receptors of Neisseria meningitidis: promising candidates for a broadly cross-protective vaccine. J Med Microbiol 1996;44: 237-243. 15. Makrides SC. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 1996;60: 512-538. Copyright 1998 Elfos Scientiae The following images related to this document are available:Photo images[ba98042b.jpg] [ba98042c.jpg]Line drawing images[ba98042a.gif] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}