 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Hacia La Primera
Década De Estudio Del Factor Mónica Bequet Romero, Omar López Ocejo Centro de Ingeniería Genética y
Biotecnología. AP 6162, CP 10600, Code Number: BA99001
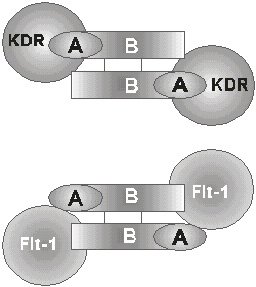
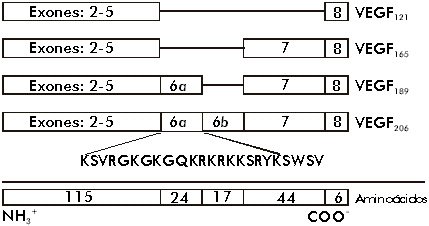
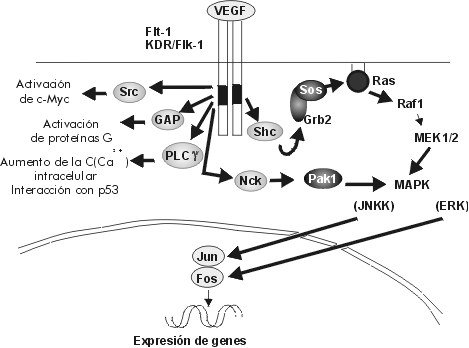
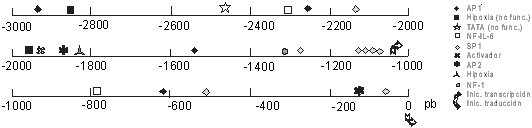
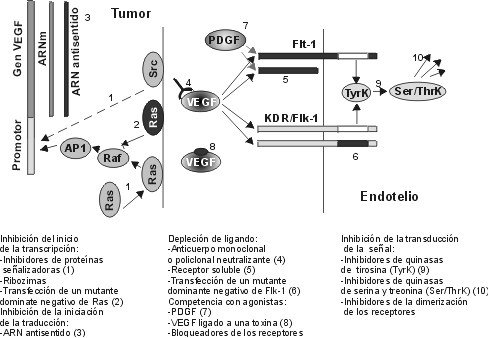
ABSTRACT The angiogenic process is related with the implantation, growth and development of tumors. One of the most important and specific angiogenic factors is the vascular endothelial growth factor (VEGF), also known as the vascular permeability factor. This molecule was first described in the late 80's and the investigations about its role in multiple processes have increased in the last years. The present review offers an overview on current knowledge about VEGF and its receptors, including a brief description of the signals that induce VEGF transcription and those generated by VEGF-receptor binding. The use of the elements involved in the signal transduction pathways as targets for anti-cancer therapies, are also summarized. Keywords: angiogenesis, Flk-1, Flt-1, KDR, VEGF, VPF RESUMEN El proceso angiogénico se encuentra estrechamente relacionado con la implantación, el crecimiento y el desarrollo de tumores. Uno de los factores angiogénicos de mayor importancia, dada su especificidad, es el factor de crecimiento del endotelio vascular (VEGF), también conocido como factor de permeabilidad vascular. Esta molécula se describió por primera vez a finales de la década de 1980, y la investigación acerca de ella se ha incrementado en los últimos años. El presente trabajo pasa revista al conocimiento actual acerca del VEGF y sus receptores, e incluye una breve descripción de las señales que inducen la transcripción del VEGF y de aquellas que son generadas por la unión del VEGF a sus receptores. También, se resumen los componentes de las vías de transducción de señales que se han explorado invitro e invivo como un blanco para las terapias antitumorales. Palabras claves: angiogénesis, Flk-1, Flt-1, KDR, VEGF, VPF Introducción El sistema vascular es esencial para el suministro de oxígeno y nutrientes, la eliminación de los desechos del metabolismo, y es el medio que facilita el acceso de los leucocitos a los tejidos de todo el organismo. Los rudimentos de este sistema se forman durante la embriogénesis al diferenciarse los angioblastos a células endoteliales (vasculogénesis). El crecimiento posterior de los vasos sanguíneos ocurre por proliferación de las células del endotelio vascular, ya formadas en respuesta a factores secretados por los tejidos aledaños (angiogénesis) [1]. La angiogénesis es un proceso esencial en diversos eventos fisiológicos como el desarrollo y remodelación de órganos en la etapa fetal, la reparación de daños y la regeneración de tejidos [2]. Cuando el crecimiento de los vasos es descontrolado, la angiogénesis se hace patológica y favorece la progresión de enfermedades neoplásicas y no neoplásicas. Entre las no neoplásicas se destacan la artritis, donde los nuevos capilares invaden el cartílago y lo destruyen, el síndrome de ceguera asociado a la diabetes y la neovascularización ocular, en las cuales los capilares ocupan el humor vítreo y ocasionan pérdida de la visión [3]. En el caso de las afecciones neoplásicas se destacan los tumores sólidos. Se plantea que estos tumores no alcanzan una talla mayor de 2-3 mm3 hasta que no se vascularizan. Durante la etapa prevascular las células de tumores y micrometástasis se replican tan rápido como aquellas de los tumores en expansión, ya vascularizados, pero sin el crecimiento de nuevos vasos, se establece un equilibrio entre la velocidad de proliferación y muerte que limita el crecimiento del tumor. Sólo al ocurrir un cambio en el fenotipo angiogénico en las células tumorales, es posible alterar el balance de factores angiogénicos y antiangiogénicos, con lo que comienza la vascularización del tumor y su crecimiento [4,5]. A la relación entre el evento angiogénico y el crecimiento, desarrollo e incluso implantación del tumor, se le ha prestado gran atención en los últimos tiempos, sobre todo desde el punto de vista de las posibilidades terapéuticas de un tratamiento antiangiogénico en la lucha contra el cáncer [6-10]. En vista de lo anterior se han dedicado grandes esfuerzos en las últimas dos décadas al estudio de todos los factores que pudieran influir, de una forma u otra, sobre el control del proceso angiogénico. Antecedentes Las primeras proteínas angiogénicas se identificaron y purificaron en la década de 1980 e incluyen, entre otros, los factores de proliferación y transformación a y b (TGF-a y TGF-b ), el factor de necrosis tumoral (TNF), la angiogenina y la prostaglandina E2 [11]. Sin embargo, estos agentes presentan poco o ningún efecto mitogénico sobre las células del endotelio vascular. Por lo tanto, se piensa que ejercen su acción angiogénica a través de la modulación de inductores directos que podrían derivarse tanto de macrófagos, como de la membrana basal [2]. En esta misma época, se describieron otros factores angiogénicos pertenecientes a la familia de los factores de crecimiento de fibroblastos (FGF) que, a diferencia de los anteriores, sí resultan mitogénicos para el endotelio vascular, pero no son específicos para éste. Los FGF inducen, además, la proliferación de un gran número de tipos celulares, lo que se piensa que promueva la angiogénesis como parte del crecimiento coordinado de los tejidos y de su reparación [12,13]. Con esto último, el hecho de que los FGF no poseen el péptido señal requerido para el transporte extracelular por la vía clásica de secreción, hace pensar que estos factores, aunque pueden ser importantes, no son los mediadores principales de la angiogénesis, pues existen múltiples evidencias experimentales de que este proceso requiere la liberación de factores difusibles [14,15]. En 1989, se describió el primer factor de crecimiento específico para el endotelio vascular, que es secretado al medio por la vía clásica. Se le denominó factor de crecimiento del endotelio vascular (VEGF) o factor de permeabilidad vascular (VPF), debido al ensayo que se usó en cada caso para monitorear la actividad biológica de las moléculas[2,16,17]. El VEGF se describió inicialmente como una proteína secretada por células tumorales, que genera un efecto de hiperpermeabilidad (con una potencia 50000 veces mayor que la de la histamina) a macromoléculas circulantes en microvasos, vénulas postcapilares primarias y pequeñas venas [8]. Por otra parte, el VEGF se ha demostrado que altera el patrón de expresión génica en las células endoteliales e induce la angiogénesis invivo [18,19]. Se ha observado, además, que el ARN mensajero (ARNm) del VEGF se expresa en tejidos normales, fundamentalmente en aquellos para los que es de vital importancia el intercambio continuo con la sangre. Por ejemplo, se demostró que, mientras la expresión de VEGF es esencial en el hígado y el páncreas, en el tejido cerebral adulto ésta se hace prácticamente nula [20]. A diferencia del estado adulto, donde la proliferación de vasos está altamente restringida a zonas como el cuerpo lúteo, en el embrión en desarrollo éste es un proceso común y vital relacionado temporal y espacialmente con la expresión del VEGF [21]. La adición del VEGF a células endoteliales en cultivo, provoca la acumulación de iones calcio en el citosol, cambios de forma, división celular y migración, todos eventos necesarios, ya sea para el aumento de la permeabilidad vascular o para el proceso angiogénico. Además, el VEGF también puede actuar como un factor de supervivencia para aquellas células que expresen los receptores para él, ya sean células endoteliales o tumorales. El ARNm del VEGF ha sido detectado en un gran número de líneas celulares tumorales tanto humanas como murinas, al igual que en tumores primarios, localizándose, en estos últimos, en la zona que rodea el área necrótica [14,22,23]. Existen numerosas evidencias que correlacionan la expresión del VEGF con el grado de malignidad de los tumores. En los glioblastomas, la expresión del VEGF es mucho mayor que en los gliomas de bajo grado [20], y en las neoplasias cervicales se observa una situación similar [24]. Actualmente, el VEGF se considera uno de los factores angiogénicos más importantes implicados en el desarrollo tumoral, de ahí que constituya un foco de atención para la terapia del cáncer [8]. Caracterización estructural del VEGF El VEGF es una molécula termoestable, resistente en medio ácido, pero lábil al tratamiento con agentes reductores [15], que es secretada como un homodímero (su forma activa) de peso molecular entre 34 y 46 kDa. Esta proteína pertenece a la familia del factor de crecimiento derivado de las plaquetas (PDGF), con el que comparte una homología en su secuencia de aminoácidos (aa) de 20% aproximadamente, relacionándose más aún (53% de homología) con el factor de crecimiento de la placenta (PlGF). Es de importancia la conservación en las tres moléculas de ocho residuos de cisteína esenciales para el establecimiento de la estructura terciaria de la proteína y para su interacción con los receptores de membrana y solubles [11,16]. Recientemente, esta familia ha sido ampliada luego del aislamiento de dos nuevos factores de crecimiento con estructuras homólogas al VEGF, designados VEGF-B y VEGF-C [25-27]. Al igual que el PDGF y otros factores de crecimiento, el VEGF también es una glicoproteína. La N-glicosilación ocurre en el residuo Asn75 y resulta esencial para mantener la estabilidad de la proteína y garantizar su secreción eficiente, pero no tiene influencia sobre la actividad biológica [28]. Para el VEGF se han descrito cuatro isoformas fundamentales que comparten entre sí una zona común en el extremo amino, diferenciándose en el extremo carboxilo. Es en el extremo amino donde se localizan los determinantes de unión para los receptores, de manera que en ella recae la actividad biológica de este factor de crecimiento. No obstante, hacia el extremo carboxilo, en dependencia de la isoforma, se encuentran o no residuos capaces de modular la interacción con los receptores. En un modelo tridimensional del dímero de VEGF (dominio N-terminal: 1-110), basado en la estructura cristalográfica del dímero de PDGFb, se observa que la cadena polipeptídica de cada monómero se encuentra compactada en dos pares de cadenas antiparalelas en conformación de hoja b plegada; seis de las cisteínas conservadas forman tres puentes disulfuro intracatenarios, mientras que las dos restantes son responsables de la unión covalente que permite la formación de la estructura homodimérica [29]. Una vez formado el homodímero, la molécula adopta una conformación elongada que expone tres lazos hacia la superficie agrupados hacia los extremos de la molécula. Por mutagénesis dirigida se ha identificado una zona de carga positiva en uno de los lazos expuestos (lazo III), que media la unión al receptor KDR/Flk-1 y que incluye los aminoácidos Arg82, Lis84 e His86, mientras que los residuos Asp63, Glu64 y Glu67, que conforman una zona de carga negativa (lazoII), resultaron ser esenciales para la unión al receptor Flt-1. En el monómero, estas zonas cargadas se encuentran alejadas entre sí, pero al producirse la dimerización se acercan espacialmente, lo que conduce a la aparición de sitios de unión para los receptores en ambos extremos de la molécula. Este último dato concuerda con la bivalencia de los factores que se agrupan en esta familia y además sugiere un posible mecanismo para la dimerización de los receptores, inducida por ligando [29] (Figura1). Figura 1. Epítopes del VEGF para la unión a sus receptores. Se describen dos sitios diferenciales para la unión a KDR (sitio A) y a Flt-1 (sitio B). El sitio A está compuesto por los residuos básicos presentes en la región 82-86, mientras que el sitio B está constituido por los residuos ácidos de la región 63-67. La composición del extremo carboxilo depende de la variante de corte y empalme del VEGF que se trate. Hasta ahora, se han descrito cuatro isoformas del VEGF resultantes del procesamiento alternativo del ARN nuclear heterogéneo (ARNnh), las cuales contienen 121, 165, 189, y 206 aa. Existen algunas evidencias de la existencia de una quinta variante que codificaría para una isoforma de 145 aa, pero esta especie no se ha identificado aún [30]. Las isoformas del VEGF se diferencian significativamente en su capacidad de unión a la heparina y heparan sulfatos los cuales parecen jugar el papel de "chaperonas", protegiendo al factor de crecimiento ante las condiciones oxidantes generadas en procesos inflamatorios y heridas donde la acción del VEGF es esencial. Esta proteína es codificada por un solo gen que comprende ocho exones, el primero de los cuales, al igual que en un gran número de proteínas secretadas, codifica prácticamente en su totalidad para la secuencia del péptido señal que viabiliza la secreción por la vía clásica. Todas las isoformas comparten extremo amino, que es codificado por los exones del 2 al 5, y además, los seis aminoácidos del extremo carboxilo codificados por el exón 8. Las diferencias antes mencionadas en cuanto a la capacidad de unión, dependen de la incorporación de los segmentos codificados por los exones 6, 7 y 8 (Figura2). Figura 2. Composición de las variantes de corte y empalme del VEGF. Los péptidos codificados por los exones del gen humano del VEGF se muestran en rectángulos (no están a escala). El número de aminoácidos para los que codifica cada uno de los exones se muestra al final. La secuencia aminoacídica representada corresponde a la denominada señal de unión a la matriz y presenta un total de 12 aa con carga positiva. La variante de 121 aa es codificada por los exones del 2 al 5 y el 8, mientras que la de 165 aa, presenta una secuencia polipeptídica catiónica adicional codificada por el exón 7, que le confiere la habilidad de unirse a la heparina y a los proteoglicanos de heparan sulfato. Este péptido de 44 aa posee nueve residuos aminoacídicos cargados positivamente (Lis,Arg), pero su estructura no concuerda con los sitios de unión a heparina descritos en la literatura. Al analizar la funcionabilidad del monómero de VEGF165, se ha observado que éste no presenta gran afinidad por la heparina comparado con el homodímero, lo cual indica que la presencia de las dos zonas de carga positiva es lo que parece conferir a esta molécula dicha facultad. La isoforma de 189 aa posee 24 aa más, codificados por la porción a del exón 6, dentro de los cuales se ha localizado una zona con una alta densidad de carga positiva (12 residuos básicos) cuya conformación concuerda con la estructura esperada para un dominio de unión a heparina, de manera que el VEGF189 posee dos de estos sitios. La variante de 206 aa, que se encuentra con muy poca frecuencia y sólo se ha identificado en una biblioteca de ADNc de hígado humano, se diferencia del VEGF189 por la presencia de 17 aa más codificados por la porción b del exón 6, presentando las mismas características en lo que a unión a heparina se refiere. La presencia, tanto en el VEGF189, como en el VEGF206, del segmento polipeptídico codificado por el exón 6a, también denominado señal de unión a la matriz, condiciona la alta afinidad de estas moléculas por los polianiones endógenos y, por lo tanto, la permanencia de éstas asociadas a la matriz extracelular, mientras que el VEGF165 y el VEGF121 son secretados en forma soluble [11,17,21] (Figura2). El hecho de que las isoformas mayores (VEGF189 y VEGF206) permanezcan asociadas a la membrana, ha conducido a la especulación de que los mismos juegan un papel en las interacciones celulares o en la transducción de la señal, lo cual se ha corroborado en parte por el hecho de que la isoforma de 189 aa aparece sobrerregulada en cultivos confluentes [22]. Generalmente, se ha aceptado que el VEGF165 es la isoforma más abundante [31], pero en un estudio reciente sobre los niveles de expresión de varias especies de ARNm del VEGF en órganos de rata, se ha demostrado que la prevalencia de una isoforma u otra depende del tipo celular y del tejido, encontrándose, por ejemplo, que en corazón y pulmón la isoforma predominante es el VEGF189 [30], mientras que en la placenta, el VEGF121 es mayoritario [28]. Las diferentes isoformas del VEGF también pueden ser procesadas por mecanismos postraduccionales, de manera que se obtienen otras isoformas también activas como ocurre en el caso de la fragmentación por plasmina del VEGF189 y el VEGF165. Con ello, se obtiene una variante de 110 aa con actividad similar a la del VEGF121 y otro fragmento con afinidad por la heparina [21]. De lo anterior puede concluirse que los procesos de corte y empalme alternativos y/o la proteólisis limitada modulan la unión del VEGF a los proteoglicanos de la matriz extracelular, controlando de esta forma la difusión desde los sitios de síntesis y determinando la extensión del almacenamiento local. Por otra parte, estos eventos generan una heterogeneidad estructural y funcional que parece explicar la variedad de respuestas biológicas y también el gradiente decreciente de respuesta que se genera desde el foco angiogénico hasta la periferia; ello aporta direccionalidad al proceso. Receptores para el VEGF Se conocen dos receptores de alta afinidad para el VEGF/VPF, que se denominan KDR/Flk-1 (receptor con dominio inserto-quinasa/quinasa de hígado fetal: homólogo murino) y Flt-1 (quinasa de tirosina del tipo fms). Estas moléculas pertenecen a los receptores de clase III del tipo quinasa de tirosina. Ambos contienen un solo dominio transmembranoso, siete dominios extracelulares del tipo inmunoglobulina y una región intracelular que contiene un dominio quinasa de tirosina truncado por una zona larga denominada inserto quinasa (60-70 aa) rica en residuos de tirosina fosforilables [32]. Además de estos dos receptores localizados a nivel de membrana, también se ha descrito una alternativa de corte y empalme del Flt-1, denominada sFlt-1, que carece del dominio carboxilo terminal y, por lo tanto, aparece soluble, de manera que al conservar sus propiedades de unión al VEGF es capaz de bloquear los efectos del mismo [11, 33]. Las secuencias aminoacídicas de los receptores KDR/Flk-1 y Flt-1 comparten una homología de 45%, pero, a pesar de ello, existen múltiples evidencias que apuntan hacia una divergencia funcional entre ellos. Por ejemplo, utilizando mutantes del VEGF relativamente selectivos para KDR o Flt-1, se ha observado que los mutantes para unión a Flt-1 desarrollan una actividad normal en cuanto a proliferación se refiere, lo cual concuerda con la falta de crecimiento en respuesta al PlGF que se une a Flt-1 y no a KDR [34]. En otros experimentos con células que expresan KDR se ha constatado que el VEGF induce cambios drásticos en la morfología celular, organización de la actina, quimiotaxis y mitogenicidad, mientras que las células que expresan el receptor Flt-1 no desarrollan estas respuestas [29]. Por otra parte, en ensayos con ratones deficientes para cada uno de los receptores en particular, se detectó que, aunque la eliminación de cualquiera de las dos moléculas conduce a la muerte intrauterina hacia los días ocho a diez de gestación, se observa un patrón diferente de anomalías en el desarrollo vascular en dependencia de qué receptor no se exprese. En embriones murinos homocigóticos para una mutación en el gen KDR/Flk-1, se aprecia un defecto en el desarrollo de células endoteliales y hematopoyéticas, ya que no se observan vasos sanguíneos ni en el embrión ni en el saco vitelino y los progenitores hematopoyéticos se encuentran severamente reducidos. En el caso de los embriones que carecen de Flt-1, se observan células endoteliales en las regiones extraembrionarias, pero el ensamblaje en canales vasculares es totalmente anormal tanto en la microvasculatura como en los grandes vasos [25,35,36]. Así, de las evidencias anteriores puede inferirse que, mientras el receptor KDR/Flk-1 es esencial para desencadenar una respuesta mitogénica, la señal vía Flt-1 es la responsable de la regulación de las interacciones de las células endoteliales entre sí y con la matriz. Además, el receptor Flt-1 resulta importante para las interacciones del endotelio con otros tipos celulares como los fagocitos mononucleares durante la respuesta inflamatoria [37]. Un acercamiento desde el punto de vista molecular indica que el receptor Flt-1 es menos fosforilado o presenta un recambio más rápido de fosfotirosinas que el observado para KDR/Flk-1, lo cual, en vista de la importancia de las fosfotirosinas para la activación de las cascadas citoplasmáticas de transducción de señales, parece indicar que ésta pudiera ser la causa de la disparidad funcional entre estos receptores [28]. Durante el desarrollo normal, los receptores del VEGF parecen expresarse durante la vasculogénesis y la diferenciación vascular desde las primeras semanas de vida del embrión, en los precursores de las células endoteliales conocidos como hemangioblastos, que se localizan en los islotes sanguíneos. Inicialmente, se pensó que la expresión de estos receptores estaba restringida al endotelio vascular, tanto en el embrión, como en el adulto, durante la angiogénesis normal y la patológica [38-41]. No obstante, se han detectado receptores para el VEGF en células derivadas de la capa muscular lisa del útero [42], en líneas celulares derivadas de cáncer de próstata, de mama y en células de carcinoma ovárico [30,35]. La expresión conjunta del VEGF y sus receptores, unido al hecho de que el VEGF es capaz de inhibir la vía apoptótica al unirse a sus receptores, da una idea de cómo el VEGF secretado por las células tumorales puede resultar importante, no sólo para mantener la vasculatura tumoral, sino también para garantizar la supervivencia de las mismas por otras vías. Recientemente, se ha demostrado que la expresión de Flt-1 y KDR aumenta en las células endoteliales asociadas al tumor durante la progresión del mismo [43]. Debido al papel de las moléculas Flt-1 y KDR/Flk-1 como mediadoras de la acción del VEGF sobre el proceso de la angiogénesis tumoral, se ha planteado la posibilidad de que éstos sean inducidos por el VEGF o, al igual que este último, por hipoxia [10,44,45]. Al igual que otros receptores para factores de crecimiento, los del VEGF, al unir su ligando, se dimerizan. Así, se activan los dominios intracelulares que presentan actividad quinasa de tirosina y de este modo se experimenta tanto autofosforilación como transfosforilación de la subunidad adyacente. De esta forma, se activa el dominio quinasa de tirosina del receptor que puede fosforilar proteínas mediadoras de la transducción de la señal, ya sean citosólicas o de membrana que presentan sitios SH2 para la unión al dominio de fosfotirosinas que se forma en la zona del inserto quinasa del receptor. Entre estas proteínas se encuentran la fosfatidilinositol-3-quinasa (PI3K), la fosfolipasa Cg (PLCg ), las quinasas MAP (MAPK), las proteínas adaptadoras como Shc y Grb2, las proteínas Yes y Fyn, las proteínas GAP (GTPasa activadora de la proteína Ras) y NcK. De estas enzimas, la PI3K y la PLCg generan una serie de segundos mensajeros como el trifosfato de inositol y el diacilglicerol, que condicionan el aumento de la concentración intracelular de Ca2+ y la activación de la proteína quinasa C (PKC), respectivamente, mientras que las proteínas GAP y NcK pueden acoplar los receptores de la superficie celular a otros mecanismos efectores [18,33]. Esto conduce, en conjunto, a la entrada de la célula endotelial en la fase S del ciclo celular [1]. El aumento de la concentración de calcio intracelular ocurre rápidamente luego de una inyección de VEGF/VPF [18] y paralelo a ello, se observan otros efectos entre los que se destaca el aumento de la permeabilidad vascular debido a la apertura de las uniones intracelulares y al cambio hacia el fenotipo fenestrado que se observa en el endotelio de capilares y vénulas [46]. Recientemente, se ha postulado que el aumento en la permeabilidad ocurre a expensas del incremento de la actividad funcional del organelo vesículo-vacuolar (VVO), que ofrece una vía por la cual, tanto el plasma, como sus proteínas, pueden salir de la circulación para entrar en los tejidos [47]. Otras investigaciones en esta área relacionan las propiedades vasoactivas del VEGF con la liberación de óxido nítrico o con la apertura de canales de potasio dependientes de ATP [44]. De manera general, el VEGF altera el patrón de activación génica de las células endoteliales, sobrerregulando los activadores del plasminógeno y de otras proteasas como la colagenasa y el factor tisular [33]. No obstante, excepto los primeros pasos de la señalización descritos anteriormente, se conoce muy poco acerca de las etapas intermedias que conducen a la activación de los genes ya mencionados [48] (Figura3). Figura 3. Modelo de transducción de la señal por los receptores del VEGF después de la dimerización inducida por el ligando. La unión del VEGF a sus receptores (Flk-1 y Flt-1) conduce a la fosforilación del dominio inserto quinasa intracelular. Esto favorece la unión de proteínas señalizadoras Src, GAP, PLCg, Shc, Nck), las cuales activan a su vez otras moléculas responsables de la transducción de la señal (c-myc, Grb2-Sos, Pak1) que continúan toda una cascada de señalización vía Ras, Raf1, MEK1/2. Esto último favorece la activación de proteínas quinasas activadas por mitógenos (MAPK), que según sean de tipo JNKK o ERK propiciarán la fosforilación y consecuente activación de los factores transcripcionales Jun y Fos, respectivamente. La activación de la transcripción a través de las proteínas Jun y Fos, conduce a la expresión de un grupo de genes que está sesgado por los demás moduladores de la transcripción que se activan y por el tipo de endotelio, condicionado a su vez por el tejido al que este se encuentre asociado. Regulación de la expresión del VEGF En la última década, ha aumentado el número de estudios acerca de la regulación de la expresión del VEGF y esto se debe al gran número de trabajos que han contribuido a esclarecer la estructura del gen y en especial de sus regiones reguladoras, incluyendo el análisis de las regiones flanqueantes 5´ y 3´. El gen del VEGF ha sido mapeado en el cromosoma 6, en la posición p21.3, por hibridación in situ, lo cual ha ayudado a esclarecer las bases genéticas de algunos síndromes congénitos que mapeaban en la banda 6p21.3 humana, como la hemocromatosis y el defecto del septo atrial tipo II [36,49]. El estudio de la secuencia promotora del gen del VEGF revela la existencia de un sitio principal de iniciación de la transcripción que se encuentra cercano a cuatro sitios de unión del factor transcripcional Sp1, con un alto contenido de GC, que, debido a la ausencia de una secuencia TATA, constituye uno de los principales elementos reguladores de la iniciación de la transcripción. Además, en la zona promotora se localizan múltiples sitios de unión para activadores de la transcripción de los tipos AP1, AP2, NF1 (factor nuclear1), conjuntamente con elementos de respuesta de tipo1 a IL-6 (unión a NF-IL6) y elementos de respuesta a hipoxia. A continuación del sitio de iniciación de la transcripción se localiza una zona que se transcribe y, sin embargo, no se traduce (5´UTR), la cual es excepcionalmente larga para este gen en particular (1038 pb). En esta zona se localizan también varios sitios de unión para factores transcripcionales y se ha llegado a postular que estos elementos cooperan sinergísticamente con aquellos ubicados en la zona promotora [17,50] (Figura4). Figura 4. Representación de la región del promotor y del 5'UTR del gen del VEGF humano. Se indican las regiones consenso para la unión de factores transcripcionales, así como otras regiones importantes para la regulación de la transcripción del gen. En esta zona se han descrito ocho sitios de unión para el factor transcripcional Sp1 (-2138 a -2133, -1277 a -1272, -1133 a -1128, -1118 a -1123, -1107 a -1112, -1091 a -1096, -513 a -518, -60 a -65), además de cuatro sitios de unión para factores tipo AP1 (-2930 a -2924, -2259 a -2265, -1528 a -1522, -614 a -620), dos para factores tipo AP2 (-1875 a -1868, -128 a -135), 2 para el factor NF-IL-6 (-2313 a -2320, -786 a -804), y un sitio consenso para la unión del factor NF-I (-1316 a -1314). También se señalan los sitios de respuesta a hipoxia funcionales (-1829 a -1782) y no funcionales (-2856 a -2847, -1958 a -1949), la secuencia TATA no funcional (-2465 a -2461), el activador (-1938 a -1838) y los sitios de inicio de la transcripción (-1038) y la traducción (0). La región 5´UTR presenta un alto contenido de GC (83%), lo cual parece ser de gran importancia para mantener la estructura secundaria del ARNm, que garantiza una traducción correcta, a diferencia de lo que sucede con el PDGF donde la región 5´UTR inhibe el proceso de rastreo ribosomal. Se ha sugerido, además, que esta estructura puede servir como un sitio interno de entrada de ribosomas (IRES), de manera que en este caso se podría observar iniciación interna aparte de la que ocurre por la vía convencional dependiente de CAP [50]. Además de lo anterior, la existencia de un codón ATG 185-187 nucleótidos antes del codón propuesto para la iniciación de la traducción, marca otro punto probable para la regulación a nivel traduccional de la expresión del VEGF [17]. El análisis de la secuencia que codifica para el VEGF (14pb) muestra homología con la del PDGF, sobre todo en la zona que determina la estructura de la porción amino terminal de la proteína, incluyendo tanto la distribución de ocho residuos de cisteína, como la organización de los intrones y exones que presentan las señales convencionales de corte y empalme. Aunque no se ha descrito un mecanismo preciso para regular la alternativa de corte y empalme que se produce en cada momento, éste debe existir a algún nivel para garantizar los niveles diferenciales de las isoformas encontradas en diferentes tejidos [30,51]. Por su parte, la región 3'UTR posee cerca de 420pb y en ella se destaca un sitio funcional para la unión de factores de respuesta a la hipoxia y que podría facilitar la estabilización del ARNm inmaduro en esas condiciones. La secuencia de poliadenilación (AATAAA) se localiza a 20 pb del sitio donde se añade la cola de poliA, la cual también resulta importante en la estabilización del ARNnh [2, 16]. Esto, junto al corte y empalme diferencial, conforma la regulación postranscripcional a la que está sometido este gen. Los elementos de unión a factores transcripcionales que se localizan tanto en el promotor como en los 5' y 3'UTR, unidos a la secuencia potenciadora descrita en la zona promotora (aproximadamente entre los nucleótidos -1938 y -1838), son la base del control más fuerte ejercido sobre este gen y que ocurre a nivel transcripcional. El número de moléculas y condiciones inductoras es realmente grande, lo cual evidencia la complejidad de la regulación de la expresión de esta proteína (Tabla1). Tabla 1. Inductores del VEGF/VPF.
La mayor parte de las citocinas y factores de crecimiento inducen el VEGF mediante la activación de la transcripción. Aunque, en general, se conocen los receptores, se desconoce la vía por la que éstos llegan a activar los factores transcripcionales que median sus efectos en la mayoría de los casos. Por ejemplo, se conoce que la IL-1b , el PDGF, el TGF-b y el EGF [52, 55, 56, 58, 60] activan AP1, y que la activación de la transcripción por TGF-a requiere de AP2 [57], mientras que la IL-6, al unirse a su receptor, media la inducción de VEGF vía NF-IL-6 [50]. Para otras citocinas, como el IFN-b y el TNF-a , el mecanismo de inducción se desconoce por completo. En experimentos recientes acerca de la inducción de esta proteína en queratinocitos se observó que el factor de crecimiento de queratinocitos (KGF) también es capaz de activar la transcripción del gen, pero de una forma más rápida, ya que es independiente de la síntesis proteica [53]. Esta última observación incluye el VEGF en la creciente familia de los genes de respuesta primaria. En general, el estudio de los oncogenes siempre marchó alejado del de los factores angiogénicos, pero la sobreexpresión o activación de las oncoproteínas Ras, Raf y Src en zonas tumorales donde se han observado aumentos en las concentraciones de VEGF, ha dado base a la investigación de la acción de estas proteínas sobre la transcripción del gen de este factor de crecimiento. Por su parte, las oncoproteínas Ras, Raf y Src forman parte de las vías normales de transducción de señales que derivan en la activación de c-fos/c-Jun, de ahí que sea posible la activación de la transcripción del gen del VEGF vía sitios AP1. De esta forma se ha señalado la activación de Ras como el mecanismo responsable del cambio hacia fenotipo angiogénico en lesiones precancerosas, a través del incremento de los niveles del VEGF [43,59,61,62]. Se ha reportado, además, que las versiones virales de estas oncoproteínas, por ejemplo, v-Src, son capaces de inducir el factor de crecimiento por el mismo mecanismo [63]. Recientemente, se ha descrito que la oncoproteína E7 codificada por el genoma del virus de papiloma humano, subtipo16 (VPH-16), interactúa y transactiva el factor transcripcional c-Jun [70], de ahí que sea probable que en la infección por este virus y en el establecimiento de transformaciones malignas mediadas por él, juegue un papel importante la inducción de ciertos genes vía AP1. También, relacionadas con muchas transformaciones malignas, aparecen mutaciones en genes supresores de tumores. Entre ellas, las más abundantes son las que ocurren en el gen que codifica la p53, cuya versión mutada parece actuar sinergísticamente con la PKC en la inducción del VEGF [64,71,72]. Recientemente, se ha descrito que ciertas mutaciones en el conocido gen supresor de tumores VHL, hacen de esta proteína un buen inductor del gen del VEGF. La versión salvaje de VHL codifica una proteína que controla la actividad de la elongina, esencial en el proceso de elongación transcripcional, y las mutaciones ocasionan la pérdida del control sobre este proceso por parte del VHL. Con ello, se acelera la transcripción de ciertos genes, entre ellos el del VEGF [56,73]. Estrechamente relacionadas con la inducción del gen del VEGF por oncoproteínas y mutantes de genes supresores, se hallan las proteínas señalizadoras como la PKC y sus activadores, los cuales aumentan la transcripción del gen vía AP1. En este caso se ha encontrado que la PKC es capaz de fosforilar a las oncoproteínas Raf, activando de esta forma el mismo mecanismo propuesto para los oncogenes. De entre los activadores de la PKC, se destaca el TPA (12-o-tetradecanoilforbol-13-acetato) (éster de forbol) que induce el VEGF en un gran número de condiciones experimentales debido a su similitud con el diacilglicerol (DAG). Este éster de forbol es capaz de aumentar la actividad AP1 también a través de los sitios de respuesta a TPA presentes en el promotor de c-Jun [74]. La hipoxia es el inductor más potente que se conoce para elevar los niveles del ARNm del VEGF invitro, considerándose el oxígeno como el regulador más importante de la expresión del gen [46]. Se plantea que este factor de crecimiento está implicado en la respuesta neovascular patológica que caracteriza las afecciones de la retina, así como, en la neovascularización de los tejidos isquémicos y tumorales [75]. La inducción en este caso es consecuencia, en parte, de la activación de sensores de oxígeno como la oxidasa de PH que, en estas condiciones, aumenta su actividad y genera especies reactivas del oxígeno estabilizadoras de la estructura secundaria del ARNm del VEGF [66]. Otros sensores de oxígeno de naturaleza hemoproteica pueden ser activados en situaciones que depletan la concentración de hierro intracelular y que, por lo tanto, conducen a un cambio conformacional en la estructura del grupo hemo asociado a la proteína [11,46]. Para este último caso, se postula que pudiera existir una regulación a nivel transcripcional al igual que ocurre con la eritropoyetina [6,18]. Por ejemplo, se ha observado una activación de NF-kB con posterior inducción de la familia AP1 de factores transcripcionales (c-fos, c-Jun, c-junB y c-junD) en estas situaciones y un aumento de la actividad catalítica de la pp60src. En particular, se ha determinado que esta activación de la pp60src desencadena la fosforilación de Raf y de quinasas reguladas por señales extracelulares (ERK) pertenecientes al grupo de las MAPK, cuyo blanco final parece ser el gen del VEGF [63]. En general, el estudio de la regulación positiva sobre la expresión y estabilidad del VEGF y de su ARNm, ha avanzado mucho en comparación con la investigación de la regulación negativa, de la cual se tiene poco conocimiento. Uno de los escasos ejemplos acerca de esta forma de regulación, lo constituye la inhibición de la liberación del VEGF por células mononucleares de sangre periférica (PBMC) luego de la adición de IL-10 [76]; de ésta no se conocen aún las bases moleculares. Terapia antiangiogénica El hecho de que el VEGF se considere actualmente como el mediador central del proceso angiogénico, ha convertido a los receptores, a la cascada de señalización y a la propia molécula en blancos importantes para el desarrollo de nuevas terapias antiangiogénicas para el tratamiento de patologías cuyo desarrollo está estrechamente ligado a un aumento en el grado de vascularización. Así, se ha planteado la inhibición o bloqueo de la molécula como un medio eficaz para suprimir su efecto [77]. De hecho, el suministro de anticuerpos neutralizantes suprime el crecimiento de tumores xenógenos implantados [78,79] y reduce el número y tamaño de las metástasis en ratones desnudos a los que se les han implantado células de carcinoma de colon humano (anticuerpo monoclonal contra el VEGF en estudios preclínicos, compañía Genentech [26]). Analizando la interacción ligando-receptor, se ha observado una inhibición de la actividad mitogénica del VEGF usando la variante soluble del receptor Flt-1 que mantiene su afinidad por el ligando y, al unirse a éste, disminuye la probabilidad de unión a los receptores de membrana que desencadenan una respuesta biológica (fragmento del receptor Flt-1 soluble en estudios preclínicos, compañía Merck [80]). El uso de un anticuerpo monoclonal contra el receptor Flk-1, y pequeñas moléculas capaces de inhibir la unión del VEGF a sus receptores o la fosforilación y, por lo tanto, la activación de los mismos, también han mostrado resultados satisfactorios en los estudios preclínicos en curso (anticuerpo monoclonal contra Flk-1:DC101, compañía ImClone Systems; pequeñas moléculas inhibidoras, compañías SUGEN y Texas Biotechnology [26]). Siguiendo esta misma línea, varios laboratorios han reportado la inhibición del crecimiento tumoral al introducir una mutación dominante negativa del receptor Flk-1 que evita la transducción de la señal al citosol [81]. Por otra parte, también se ha visto que como resultado de la fusión de una toxina (DT385) al VEGF, se obtiene un conjugado (VEGF165 - DT385) efectivo en la inhibición del crecimiento invitro de las células endoteliales y de la angiogénesis invivo, en este caso resulta importante la posibilidad que ofrece el VEGF, no sólo de dirigir la respuesta hacia el endotelio tumoral, sino más específicamente hacia sus células en estado proliferativo [41] (Figura5). Figura 5. Alternativas terapéuticas dirigidas a minimizar la actividad angiogénica del VEGF. Otra de las vías fundamentales para inhibir la respuesta angiogénica generada por el VEGF es el uso de inhibidores de la transducción de la señal, ya sea de la que activa la transcripción del gen o la que se desencadena por la unión del propio VEGF a sus receptores específicos. En esta área se han obtenido buenos resultados en la inhibición de la angiogénesis en ratas, usando los inhibidores de quinasas tirosinas LavedustinA [82] y Genistein [44,63]. Por otra parte, dada la importancia de oncogenes como ras en la activación del VEGF, se han usado varios procedimientos para reducir su expresión o afectar su funcionamiento, incluyendo, entre otros, el uso de ribozimas [83,84], mutantes dominantes negativos de Ras [85,86] e inhibidores de la farnesilación de la proteína Ras, como por ejemplo, el L739 y el L749 [61,87, 88], y los péptidos miméticos benzodiazepínicos [89]. Este último método evita la incorporación de Ras a la membrana y con ello, la transducción de la señal, observándose una supresión de la producción del VEGF/VPF invitro. También se encuentran en estudios preclínicos oligonucleótidos para inhibir la expresión del VEGF en células tumorales (ARN antisentido del VEGF, compañía Hybridon [26]), que han mostrado buenos resultados en la inhibición de la neovascularización inducida por isquemia en la retina. Al VEGF, además de sus múltiples funciones, se le atribuye la inducción de ciertos genes de supervivencia (bcl-2 y la integrina a vb 3) relacionados con la regulación de la apoptosis, lo que hace que la terapia antiangiogénica dirigida contra este factor pueda conducir a la muerte programada de las células endoteliales, a la desintegración de los vasos sanguíneos y a una subsecuente regresión del tumor [62]. Aunque el entusiasmo con respecto a la terapia antiangiogénica parece justificado, es necesario reconocer que no es la "bala mágica" esperada y que pueden surgir una serie de problemas al tratar de implementar un tratamiento para el cáncer basado en la angiogénesis [48,90,91]. Entre estos problemas, se destacan las dificultades tecnológicas en los estudios preclínicos, dados por la diferencia entre la vasculatura de los tumores humanos y murinos, ya que al ser estos últimos más "jóvenes" e "inmaduros", son más vulnerables al tratamiento antiangiogénico. También es de destacar el supuesto hecho que ésta sea una terapia de baja toxicidad y que impliqua un tratamiento prolongado. Sin embargo, se conoce que la expresión del VEGF y sus receptores es fundamental en ciertos órganos, de manera que su ausencia podría acarrear efectos secundarios cuya magnitud debe ser evaluada. Un ejemplo de lo anterior podría observarse al eliminar o bloquear el receptor KDR/Flk-1, lo que puede conducir a la diabetes, pues el sistema VEGF-KDR es esencial para mantener el endotelio fenestrado en los islotes pancreáticos. Por otra parte, se conoce que el desarrollo del tumor depende de la neovascularización, pero esta dependencia está condicionada por el tipo de tumor y por el estadio en que se encuentre, ya que se ha visto que a medida que pasa el tiempo las células tumorales se hacen más independientes de los factores de crecimiento y, por lo tanto, de la irrigación sanguínea [9, 20]. Teniendo en consideración lo antes expuesto, se impone continuar el estudio de factores angiogénicos como el VEGF para completar el conocimiento acerca de la regulación de su expresión, así como la de sus receptores, considerando las cascadas de señalización, todo lo cual podría aportar un blanco más perfecto para dirigir una terapia antiangiogénica que, combinada con otras drogas, contribuya al desarrollo de una estrategia que resulte más eficiente en el tratamiento del cáncer. Agradecimientos Los autores agradecemos al doctor Manuel de J. Araña por la provechosa revisión y discusión de este trabajo. Referencias
Copyright 1999 Elfos Scientiae The following images related to this document are available:Photo images[ba99001e.jpg] [ba99001b.jpg] [ba99001a.jpg] [ba99001d.jpg] [ba99001c.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}