 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Detection Of Deoxyribonuclease Activities Using A Pcr-Generated Radiolabeled Dna Substrate *Frank Miranda,1,2 Luis E Trujillo,2 Félix Dafhnis,2 Enrique Pérez,2 José E Brito2 *Corresponding author 1 Centro de Ingeniería Genética y Biotecnología. AP 83, Zona 2, Sancti Spíritus 62500, Cuba.2Centro de Ingeniería Genética y Biotecnología. Ave 31 entre 158 y 190. AP 6162, CP 10600, Cubanacán, Ciudad de La Habana, Cuba. Telf: (53-7) 21 8466; Fax: (53-7) 33 6008; E-mail: IPMlab@cigb.edu.cu Code Number: BA99007
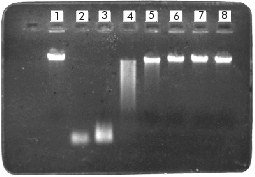
ABSTRACT Here we evaluated a radioactive DNA substrate for the efficient detection of deoxyribonuclease activities in final preparations of DNA restriction and modifying enzymes and other biological reagents as acetylated BSA. The substrate was obtained by radioactive PCR and it was found to be more sensitive in the detection of deoxyribonuclease activities (more than 10-5 units of a commercial DNAse I in 20 min of reaction) than the non-radioactive substrates commonly used for these purposes. Keywords: deoxyribonuclease activity, PCR, quality control RESUMEN En este trabajo se evalúa un sustrato radiactivo creado para la detección eficiente de actividades de desoxirribonucleasas contaminantes de las preparaciones finales de enzimas de restricción y modificación del ADN y otros reactivos biológicos como la BSA acetilada. Este sustrato obtenido mediante PCR radiactivo resultó ser más sensible en la detección de desoxirribonucleasas (más de 10-5 unidades de ADNasa I comercial en 20 min de reacción) que los sustratos no marcados radiactivamente utilizados comúnmente para estos propósitos. Palabras claves: actividad de desoxirribonucleasa, control de calidad, RCP Introduction DNA restriction and modifying enzymes are produced by different microorganisms and are widely used in molecular biology. The isolation and purification procedures must ensure the total elimination of non-specific activities that could be present in final preparations of these enzymes. Current analytical quality control methods for the detection of undesirable deoxyribonuclease activities in final preparations of biological reagents such as restriction and modifying enzymes, acetylated BSA, etc, involve the use of different DNA substrates [1-3]. In some cases, a 16 h overincubation at 37 ºC of linear or superhelical DNAs with aliquots of the assayed preparation are used for these purposes. Here we used the polymerase chain reaction (PCR) [4] for the obtainment of a highly-specific activity amplification product (7.0 x 106cpm/ng) by adding a labeled nucleotide [5-7]. This amplification product was used as a deoxyribonuclease substrate for quality control purposes. Yields of 100 ng of synthesised DNA and 65-70% incorporation of the radiolabeled nucleotide were steadily obtained as judged by the values of the coefficient of variation (CV) < 8%. Materials and Methods Radioactive PCR was performed using a template of 220 bp, originated from l DNA, with no restriction sites for 35 commonly used type II restriction endonucleases. Two synthetic primers of 16nt (5'CCGTTG-AATGGGCGGA3'; T = 52 ºC) and 20nt (5'ATAAGG-GTGTTGCGCTGCTT3'; Tm=60ºC) with complete homology to the 5' OH ends of each strand in the target DNA were used. PCR reactions (100mL) contained: 150 pmol of each forward and reverse primer, 250 pmol of each unlabeled deoxynucleotide, 50 mCi of [a-32P] dATP (3,000Ci/mmol; Amersham), 1 mg of l DNA (4.5 ng of the 220bp template DNA), 10 mL of the PCR reaction buffer [10 mM Tris-HCl (pH 9.0 at 25ºC), 50 mM KCl, 2.5 mM MgCl2 and 1% Triton X-100] and 2.5 units of Taq DNA polymerase (Enzibiot, Cuba). To circumvent water evaporation, 35 mL of mineral oil were added to each reaction tube. After an initial 5 min denaturation step at 95ºC, 10 cycles of 95ºC for 30 s, 40ºC for 60 s, and 72 ºC for 30 s were performed in a Thermal Cycler (Perkin Elmer Cetus, USA). Nicks columns (Pharmacia, Sweden) were used to separate free nucleotides from the radiolabeled 220 bp-long amplification product. By trichloroacetic acid (TCA) precipitation [8,9] and polyethyleneimine (PEI)-cellulose thin-layer chromatography [1], the incorporation percentage of the labeled nucleotide in the PCR-synthesised DNA was measured. For the sensitivity evaluation of this substrate, we incubated different amounts of commercial DNAse I (Boehringer Mannheim, Germany) from 1 to 10-5 units (10-fold consecutive dilution) for 20 min at 37ºC with 1ng of the generated PCR radiolabeled fragment, in the appropriate reaction buffer [40 mM Tris-HCl (pH7.9 at 25ºC), 10 mM NaCl, 6 mM MgCl2, 10mM CaCl2] and 20 mL as the final reaction volume. After incubation, 5 mL of each reaction were spotted on thin-layer chromatography PEI cellulose (Merck, Germany) strips and run until the mobile phase (HCl 2.0N) reached the top. After drying, all strips were exposed 2 h at -70 ºC (for signal development) using an Hyperfilm-MP (RP-1675; Amersham, UK) with an intensifying screen. Agarose gels to 0.8% for DNA electrophoresis [8], were prepared in buffer Tris acetate supplemented with ethidium bromide (10 mg/mL). Results and Discussion Radioactivity signals at the bottom of strips (origin) were integrated and plotted by densitometrical scanning as displayed by Figure 1 (A and B). The non-degraded DNA substrate remains at the application point where signal intensity appears to be higher with a decreasing enzyme concentration. Substrate degradation for all the assayed dilutions of DNAse I is evident since optical density at the negative control signal was greater than those corresponding to the enzyme dilutions as judged by the same figure. Figure 1. A: autoradiography results after incubation at 37ºC for 20 min of different units of DNAse I with 1 ng of the PCR-generated DNA substrate, and performance of thin-layer chromatography in PEI cellulose strips. Lane1: negative control (no enzyme added); lane2: positive control (10 units of DNAse I); lanes 3-8: 1 to 10-5 units of DNAse I (10-fold consecutive dilution). B: densitometric analysis of Figure 1A. The CV for this procedure was < 8%. C:stability of the PCR-generated substrate under different buffer conditions, incubated 1h at 37ºC. Lane 1: negative control; lane 2: DNAse I reaction buffer; lane 3: NEB-1 reaction buffer; lane 4: NEB-2 reaction buffer, and lane 5: NEB-3 reaction buffer. The CV for this procedure was < 5%. Substrate stability was tested under different salt concentration conditions. Incubations of 1 ng of the radiolabeled DNA with four different restriction enzyme reaction buffers were carried out for 60min at 37ºC. Figure 1C shows that there was no substrate degradation due to the instability caused by salt concentrations within the buffers assayed. DNAse I activity was also tested in the four restriction buffers and we found no differences in its activity in each case (results not shown). Figure 2 shows that under the same experimental conditions, using equivalent amounts (pmols of DNA) of a non-radiolabeled linear DNA substrate (l DNA) commonly used by all the leader suppliers of restriction enzymes for contaminant detection, degradation becomes relevant only in the lane corresponding to unit 1 of DNAse I. This procedure, using non-radiolabeled DNAs showed to be less sensitive for deoxyribonuclease detection when a short incubation period (20min) at 37ºC was used. When non-radiolabeled DNA is employed, incubation periods are about 12 to 24 h and the technique becomes very time-consuming. Figure 2. Linear DNA (l DNA, 48.5 kbp) incubated with different amounts of DNAse I. Lane 1: negative control (l DNA with no enzyme added, lanes 2 and 3: positive controls (lDNA incubated with 10 and 5 units of DNAseI), lanes 4-8: l DNA incubated with 1 to 10-5 units of DNAse I (10-fold consecutive dilution). According to these results, we conclude that this PCR-generated radiolabeled substrate allows detection of very low levels (10-5 units) of a commercial deoxyribonuclease in a shorter reaction time, however, further titration must be carried out to determine the lowest detection levels. The complete assay would take 5 h (including PCR reaction, DNA purification, reactions setting and exposure time). Enough substrate to test 50-70 samples for the presence of contaminant deoxyribonuclease activities could be obtained with only one PCR reaction. Other evaluation studies for contaminants detection comparing the PCR substrate with those recently used by some leading producers of restriction enzymes must be carried out, but using final preparations of these biological reagents instead of the commercial DNAses. With this method, we could evaluate the real effect and the final cost of the procedure to be introduced as a control technique. References
Copyright 1999 Elfos Scientiae The following images related to this document are available:Photo images[ba99007b.jpg] [ba99007a.jpg] |
| |||||||||

{kind=link}
{kind=link}