 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
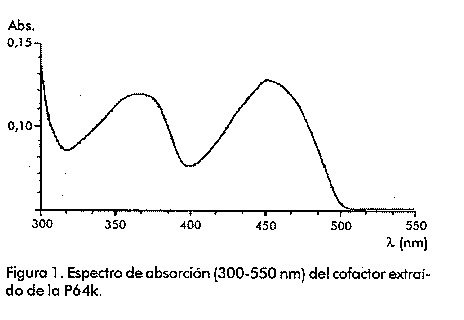
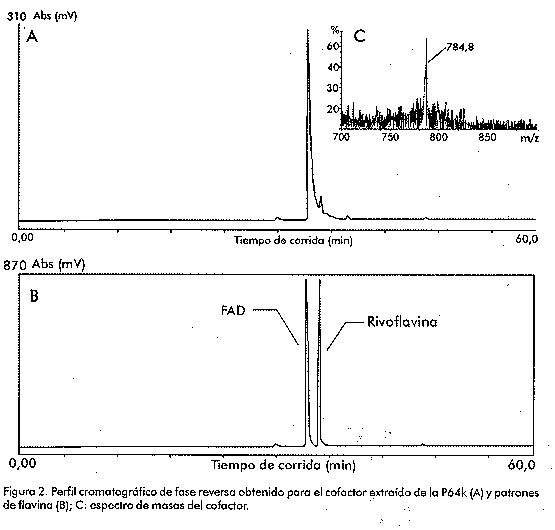
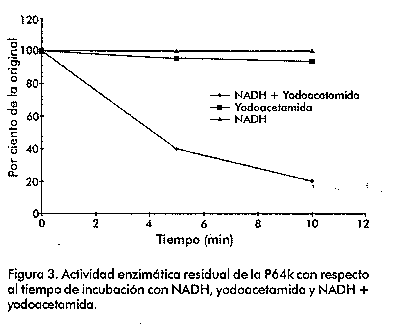
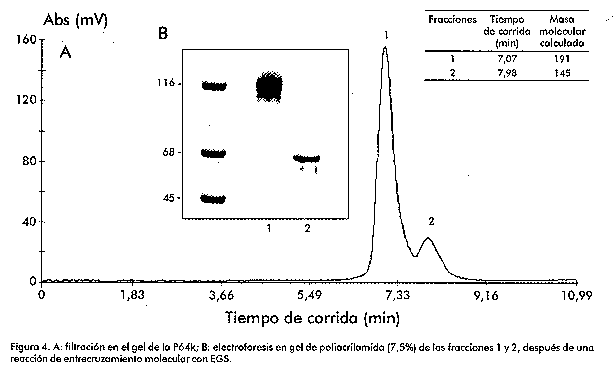
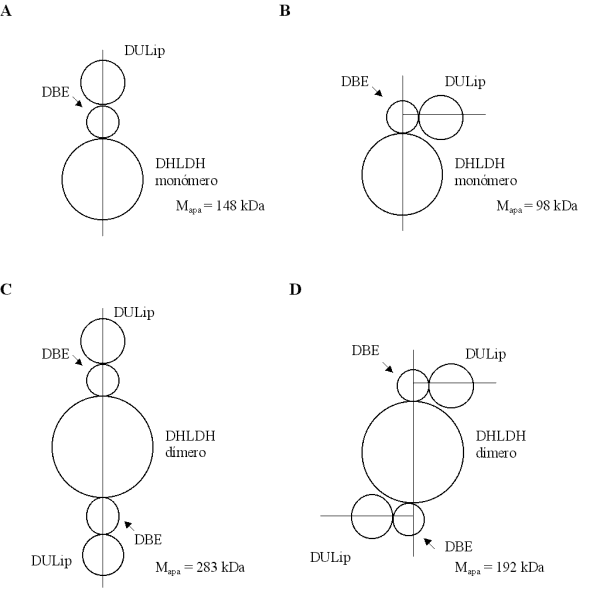
Biotecnología Aplicada 1999;16:83-87 Caracterización estructural y funcional de la proteína recombinante P64k de Neisseria meningitidis @ Raúl Gómez,1 Joel Madrazo,1 Javier González,1 Glay Chinea,1 Alexis Mussachio,2 Armando Rodríguez,1 Gabriel Padrón1 1División de Química-Física,
2División de Vacunas. Centro de Ingeniería
Genética y Biotecnología. Code Number: BA99014 Abstract The recombinant protein P64k from the bacterium Neisseria meningitidis has been identified as a dihydrolipoamide dehydrogenase. The enzyme catalyses the stoichiometric oxidation of dihydrolipoamide by nicotinamide adenine dinucleotide, involving a flavin adenine dinucleotide as cofactor. Chemical modification with iodoacetamide indicated the involvement in catalysis of a reversibly reducible disulphide bond. Migration of cross-linked protein in polyacrylamide gel electrophoresis showed that the protein is a functional homodimer. Gel filtration chromatography showed the P64k protein to migrate beyond the expected molecular weight. This abnormal behaviour is discussed taking into account the structure-function relationship. Keywords: cross-linking, dihydrolipoamide-dehydrogenase, flavoprotein, Neisseria meningitidis Resumen La proteína recombinante P64k de la bacteria Neisseria meningitidis, ha sido identificada como una dihidrolipoamida-deshidrogenasa. La enzima cataliza la oxidación estequiométrica de la dihidrolipoamida por el dinucleótido de adenina y nicotinamida, con el dinucleótido de adenina y flavina como cofactor. La modificación química con yodoacetamida indicó la participación, en la catálisis, de un puente disulfuro reversiblemente reducible. La migración de la proteína (posterior a los ensayos de entrecruzamiento molecular) en electroforesis sobre geles de poliacrilamida, demostró que la misma es un homodímero funcional. La cromatografía de filtración en gel evidenció que la P64k migra más allá del peso molecular esperado. Esta conducta anómala se discute teniendo en cuenta la relación estructura-función. Palabras claves: dihidrolipoamida-deshidrogenasa, entrecruzamiento molecular, flavoproteína, Neisseria meningitidis Introducción La bacteria gramnegativa Neisseria meningitidis es responsable de uno de cada tres casos de meningitis de tipo bacteriana en el mundo, siendo la especie humana su hospedero natural [1]. Las cepas de esta bacteria están subdivididas en tipos inmunológicos de acuerdo con la presencia de epítopes específicos correspondientes a proteínas de la membrana externa [2]. Una de las vías para la obtención de un preparado vacunal de amplio espectro de protección, es la identificación de proteínas altamente conservadas en todas las cepas, capaces de inducir anticuerpos inmunológicamente activos (anticuerpos bactericidas). La proteína P64k (Mr = 64 kDa) se encuentra presente en la membrana externa de las cepas mayormente patogénicas de N. meningitidis y es capaz de inducir anticuerpos bactericidas en su hospedero natural [3]. Con el propósito de crear una vacuna compuesta por subunidades recombinantes, el gen que codifica la P64k fue clonado y expresado en Escherichia coli [4]. Esta proteína contiene 599 residuos de aminoácidos, incluyendo siete residuos de cisteína. De su secuencia, 80% posee una elevada homología estructural con la dihidrolipoamida-deshidrogenasa (DHLDH) y el 20% restante con un dominio lipoilo [5]. Las DHLDH son flavoproteínas que forman homodímeros en un amplio número de complejos multienzimáticos como el de la piruvato-deshidrogenasa y el de la 2-oxoglutarato-deshidrogenasa. En estos complejos multienzimáticos se les conoce como E3 [6-8]. También, las podemos encontrar asociadas a la membrana citoplasmática como en el caso de la archibacteria Termoplasma acidophilum [9], el eucariota Trypanosoma brucei [10] y la eubacteria E. coli [11]. Los dominios lipoilo son elementos estructurales que forman parte de las dihidrolipoamida-acetiltranferasas. Estos elementos estructurales presentan el ácido lipoico unido covalentemente a la cadena lateral de un residuo de lisina. Las dihidrolipoamida-acetiltranferasas son otras de las proteínas que constituyen estos complejos multienzimáticos [12]. Es importante señalar que, aunque la presencia de un dominio lipoilo es una rareza en las DHLDH, ya se han encontrado casos similares en otros organismos, como las extraídas de Alcaligenes eutrophus [13], Clostridium magnum [14] y Mycoplasma capricolum [15]. Las DHLDH catalizan la oxidación reversible del dinucleótido nicotinamida adenina, forma reducida (NADH) por lipoamidas o ácidos lipoicos [16]. Durante la reacción, el NADH reduce al dinucleótido de adenina y flavina (FAD), con una posterior transferencia de electrones hacia el puente disulfuro catalítico y, de esta manera, se forma un complejo de transferencia de carga [17]. En el presente reporte, se demuestra que esta proteína recombinante posee actividad enzimática DHLDH; además, se hace una caracterización en cuanto a la naturaleza química de su cofactor y su estructura cuaternaria. Materiales y Métodos Reactivos La riboflavina, la yodoacetoamida, la DL-lipoamida y el FAD, se obtuvieron de Sigma Chemicals (St. Louis, Missouri, Estados Unidos); el etilenglicol-bis-(succinimidil-succinato) (EGS) de Pierce Chemical Co. (Rockford, Estados Unidos); los dinucleótidos nicotinamida adenina, forma oxidada (NAD+), NADH y fosfato nicotinamida adenina, forma oxidada (NADP+) de Boehringer Mannheim (Alemania); y los patrones de proteínas para cromatografía (rango 13,5-670 kDa) y electroforesis (rango 45-200 kDa) de BioRad (California, Estados Unidos). La dihidrolipoamida se sintetizó mediante la reducción de la lipoamida con borohidruro de sodio, según Reed [18]. El resto de los reactivos empleados son de calidad analítica y están disponibles comercialmente en Merck (Darmstandt, Alemania). Purificación de la proteína recombinante P64k Las células de E. coli que expresaban la P64k se lisaron en una prensa francesa a 1 500 kg/cm2. Los contenidos de ADN y proteínas contaminantes se eliminaron parcialmente mediante precipitación con sulfato de estreptomicina 1% y sulfato de amonio a 40% de saturación, respectivamente. El precipitado se separó por centrifugación a 11 000 xg durante 30 min a 4°C. El sobrenadante se aplicó en una columna cromatográfica empaquetada con una matriz de interacciones hidrofóbicas TSK-Butilo (Pharmacia, Suecia), previamente equilibrada con Tris 20 mM, pH 7,2 y NaCl 0,15 M. La P64k se eluyó con un gradiente decreciente de (NH4)2SO4 de 40 a 0% de saturación. Los productos de degradación se eliminaron mediante cromatografía de filtración en gel (CFG), empleando una columna S-200HR (Pharmacia, Suecia) equilibrada con fosfato de sodio 20 mM y NaCl 0,15 M, pH 7,5. La concentración de proteínas se determinó por el método de Bradford [19], utilizando albúmina de suero bovino como estándar. Ensayo enzimático para la determinación de la actividad dihidrolipoamida-deshidrogenasa La reacción se llevó a cabo a 25 °C en 1 mL de tampón que contenía: fosfato de sodio 150 mM, ácido etilendiaminotetraacético (EDTA) 2 mM, NaCl 50 mM, NAD+ 1 mM y dihidrolipoamida 0,4 mM, pH 7,4. Las mediciones de los valores de absorbancia se realizaron en un espectrofotómetro Hitachi 330 de doble haz de luz y paso óptico de 1 cm. La cinética se siguió por el incremento de la absorbancia a 340 nm. Los valores de actividad específica se expresaron en micromoles de NADH producidos/min por miligramo de proteínas. Modificación química con yodoacetamida La P64k (5 mg/mL) se hizo reaccionar con yodoacetamida 1 mM en presencia o ausencia de NADH 0,5 mM. En cada caso, la reacción se incubó durante 5 o 10 min a 25 °C en fosfato de sodio 150 mM y EDTA 2 mM, pH 7,4. A la proteína resultante se le determinó su actividad específica. Extracción y purificación del grupo prostético La proteína se trató con hidrocloruro de guanidina 6 M y se precipitó con sulfato de amonio a 80% de saturación. El precipitado se separó mediante centrifugación a 10 000 xg durante 10 min a 4 °C. El sobrenadante se aplicó en una columna de cromatografía líquida de alta resolución (10 x 30 mm) empaquetada manualmente con matriz de fase reversa C18 (Merck, Alemania). Posteriormente, el cofactor se eluyó con etanol 50%. El producto final se liofilizó para su posterior caracterización por cromatografía líquida y espectrometría de masas. Medición por espectrometría de masas Los espectros de masas se obtuvieron en un espectrómetro de masas de doble enfoque JEOL JMS-HX110HF, operado con el sistema de adquisición de datos JEOL JMA-DA 5 000. La muestra fue ionizada mediante bombardeo con átomos rápidos a un voltaje de aceleración de 4 kV. Los iones secundarios que se generaron de la colisión fueron acelerados a un voltaje de 10 kV. La resolución empleada fue 1 000. El cofactor se disolvió en una matriz de glicerol y la medición se realizó en el modo negativo. Los iones fueron acelerados posteriormente a -20 kV y detectados en el multiplicador electrónico secundario a -1,6 kV. Reacción de entrecruzamiento molecular y electroforesis en gel de poliacrilamida La P64k (6,7 mg/mL en fosfato de sodio 20 mM, NaCl 0,15 M, pH 7,5) se hizo reaccionar con EGS 1 mM disuelto previamente en dimetilsulfóxido a temperatura ambiente, durante 30 min. Para detener la reacción, se añadió Tris (concentración final 30 mM) y se incubó a temperatura ambiente durante 15 min. El producto se aplicó en un gel de poliacrilamida (7,5%) y la electroforesis se realizó por el método de Laemmli [20]. Caracterización por cromatografía líquida Los experimentos de cromatografía se realizaron en un sistema de alta resolución (Pharmacia-LKB, Suecia). El registro de la señal se hizo a 280 nm mediante un detector de longitud de onda variable (Knauer, Alemania). La cromatografía de fase reversa se realizó en una columna analítica C18 VYDAC 218TP5215 (California, Estados Unidos) utilizando como solución de elución acetato de amonio 0,2 M (pH 7), metanol 13% como solución A, y metanol 100% como solución B. El gradiente utilizado fue de 0 a 100% de solución B durante 60 min a 25 °C. Las separaciones mediante filtración en gel se realizaron en una columna analítica TSK-G3000SW (TOSOHAS, Japón) equilibrada con fosfato de sodio 20 mM como tampón y NaCl 0,15 M, pH 7. Para las determinaciones de peso molecular, la columna se calibró con patrones de proteínas para cromatografía (rango 13,5-670 kDa). Cálculo del radio de giro para modelos simplificados de la proteína P64k La forma de los polímeros influye en su migración en cromatografía de filtración en gel [21]. Polímeros de igual peso molecular pero de formas diferentes, tienen distintos tiempos de retención. En general, la retención de polímeros de formas diferentes se analiza comparando sus respectivos radios de giro (Rg). El radio de giro para un cuerpo rígido homogéneo se define por la fórmula siguiente: Rg = (åNX2i/N)1/2 donde: N: número de elementos de masa Xi: distancia del elemento de masa i al centro de masa del cuerpo En el caso de una esfera, la fórmula anterior se reduce a: Rg = 0,78 R donde: R: radio de la esfera La proteína P64k está formada por tres dominios estructuralmente independientes: el dominio de unión del grupo lipoilo (DUL), de aproximadamente 80 residuos de aminoácidos, el dominio brazo espaciador (DBE), de aproximadamente 30 residuos, y el dominio DHLDH, de 484 residuos [5]. Como primera aproximación, un monómero de P64k se consideró como tres esferas, cada una con un volumen proporcional a la masa del dominio correspondiente. Las dimensiones de las esferas están dadas por la relación siguiente: Ri µ (Mi)1/3 µ (Ni)1/3 donde: Ri: radio de la esfera correspondiente al dominio i Mi: masa del dominio i Ni: número de residuos del dominio i El dímero de la P64k se forma por la dimerización del dominio DHLDH. Según muestra la estructura tridimensional de este dominio [22], el dímero posee un eje de simetría rotacional C2. Por lo tanto, el dímero de la P64k se consideró como cinco esferas: una central correspondiente al dímero DHLDH y el resto correspondiente al DUL y DBE de ambas cadenas, los cuales están relacionados por un eje de simetría C2 que pasa por el centro de masa del dominio DHLDH. En dependencia de la posición relativa del DUL con respecto al dominio DHLDH, tanto el monómero como el dímero pueden formar estructuras abiertas (elongadas) o compactas. Teniendo en cuenta lo descrito anteriormente, el radio de giro para cada modelo se calculó a partir de la ecuación siguiente Rg = {[åiNi(R2gi + ri2)]/(åNi)}1/2 donde: Ni: número de residuos del dominio i Rgi: radio de giro de la esfera correspondiente al dominio i ri: distancia entre el centro de masa de la esfera del dominio i y el centro de masa global del modelo específico El valor de ri se puede calcular a partir de consideraciones geométricas simples en dependencia de la estructura específica de cada modelo. Mientras mayor sea el radio de giro de la proteína, ésta migrará como una molécula esférica de mayor masa. La masa aparente correspondiente a cada modelo es la siguiente: Mapa = (Rgmod/Rgo)3Mo donde: Rgmod: radio de giro calculado para cada modelo Rgo: radio de giro calculado para una proteína esférica de Mo = 64 kDa, para el monómero, y de Mo = 128 kDa para el dímero. Resultados y Discusión Presencia de FAD en la proteína P64k El cofactor extraído de la P64k presentó un espectro de absorción con características similares a los compuestos análogos de flavina (Figura 1), al mostrar máximos a 375 y 450 nm [23]. Para identificar la naturaleza química de este grupo prostético, se comparó, mediante cromatografía líquida de fase reversa, su tiempo de retención con respecto a dos patrones (FAD y riboflavina). El tiempo de retención del cofactor (Figura 2A) coincidió con el del FAD (Figura 2B) y su análisis por espectrometría de masas (Figura 2B) demostró concluyentemente la presencia de FAD en la P64k, al coincidir el valor m/z (784,8) con el teórico esperado (784,6). Estos resultados permiten identificar la P64k como una flavoproteína. Figura 1. Espectro de absorción (300-550 nm) del cofactor extraído de la P64k. Figura 2. Perfil cromatográfico de fase reversa obtenido para el cofactor extraído de la P64k (A) y patrones de flavina (B); C: espectro de masas del cofactor. Actividad enzimática de la proteína P64k La actividad específica encontrada en la proteína recombinante P64k fue 76 U/mg. Durante el ensayo, la velocidad de producción de NADH fue directamente proporcional a la concentración de proteínas. La P64k resultó ser específica para el NAD+: cuando este cofactor se sustituyó por el NADP+, no se detectó la actividad de la enzima. El mecanismo enzimático de las DHLDH normalmemente involucra formaciones alternativas de un puente disulfuro catalítico [24]. Este fenómeno fue empleado para establecer que la actividad enzimática observada corresponde a una verdadera DHLDH. Los grupos sulfihidrilo producto de la reducción del puente disulfuro con NADH, se bloquearon mediante la reacción con yodoacetamida. En concordancia con el mecanismo enzimático, se produjo una rápida inactivación de la enzima en presencia de NADH y yodoacetamida (Figura 3), mientras que la yodoacetamida por sí sola no causó una pérdida considerable de la actividad. Estos resultados permiten identificar la P64k como miembro de la familia de las DHLDH. Figura 3. Actividad enzimática residual de la P64k con respecto al tiempo de incubación con NADH, yodoacetamida y NADH + yodoacetamida. Estado de agregación de la P64k El estado de agregación de la proteína se determinó mediante CFG y electroforesis desnaturalizante en geles de poliacrilamida. El perfil cromatográfico (Figura 4A) mostró dos fracciones cuyos tiempos de elución correspondieron a especies de talla muy superior (145 y 191 kDa) al peso molecular de la P64k (64 kDa). Por el contrario, la migración electroforética de ambas fracciones luego de reaccionar con EGS, indicó que ellas corresponden al dímero y al monómero, respectivamente (Figura 4B). En ensayos de actividad, se observó que sólo la especie dimérica resultó ser funcional. Figura 4. A: filtración en el gel de la P64k; B: electroforesis en gel de poliacrilamida (7,5%) de las fracciones 1 y 2, después de una reacción de entrecruzamiento molecular con EGS. Efecto de la forma de la proteína en los tiempos de retención determinados por CFG La contradicción aparente entre la masa molecular del monómero y del dímero, y las estimadas a partir de los tiempos de retención en CFG, pudiera derivarse de la forma de la proteína. Para estimar el efecto de la forma de la proteína en su migración en CFG, se construyeron modelos simplificados de la P64k (Figura 5) y se calcularon los radios de giro correspondientes a cada modelo, así como su respectiva masa aparente (la masa de una proteína globular con un radio de giro similar al del modelo). El monómero de la proteína está formado por tres dominios estructuralmente independientes [5]. Cada dominio se consideró como una esfera de volumen proporcional a su masa. En los modelos del dímero de la P64k, los dominios diméricos de la DHLDH se consideraron como una única esfera con un volumen correspondiente a la masa del dímero, ya que el análisis de la estructura tridimensional del dímero de la DHLDH indica que éste es más globular que dos esferas "monoméricas" contiguas (datos no mostrados). Figura 5. Masas teóricas aparentes (Mapa) correspondientes a diferentes modelos simplificados de la proteína P64K monomérica (A y B) y dimérica (C y D). DUL, dominio de unión de la lipoamida; DBE, dominio brazo espaciador; DHLDH, dominio dihidrolipoamida-deshidrogenasa. A y C, modelos elongados de la proteína. Los centros de masa de los dominios DUL, DBE y DHLDH están situados en una misma recta. B y D, modelos compactos, los centros de masas de los dominios DUL y DBE definen una recta perpendicular a la definida por los dominios DBE y DHLDH. Como muestra la Figura 5, las masas aparentes calculadas para los modelos difieren significativamente de las masas calculadas a partir de la secuencia aminoacídica, lo que indica que, efectivamente la estructura multidominio de la proteína P64k puede explicar su migración anómala en CFG. La migración del monómero se ajusta mejor a un modelo elongado de la proteína (Figura 5A), mientras que el dímero se describe mejor mediante un modelo compacto (Figura 5D). Esto último es consistente con la función biológica de la proteína. El dímero es la conformación activa de la proteína y el sustrato del dominio DHLDH es una molécula de ácido lipoico unida covalentemente al residuo K48 del DUL. En un modelo compacto, como el que se muestra en la Figura 5D, la distancia entre el DUL y el dominio DHLDH es menor en comparación con el modelo elongado, lo que permite un acceso más fácil de la lipoamida (sustrato) al sitio catalítico del dominio DHLDH (enzima). Conclusiones La identificación del grupo prostético y la demostración de la actividad catalítica, permiten afirmar que la proteína P64k pertenece a la familia de las DHLDH. Esta conclusión está en correspondencia con la similitud encontrada a partir de la comparación de la secuencia aminoacídica de esta proteína con la de las DHLDH [5], y con los resultados obtenidos de la estructura de este dominio [22]. Por otro lado, se demostró que la P64k es enzimáticamente activa en su estado dimérico. La estructura multidominio de esta proteína explica su migración anómala en CFG. La migración del monómero se ajusta a un modelo elongado de la proteína, mientras que el dímero se describe mejor mediante un modelo compacto. Referencias 1. Devoe IW. The meningococcus and mechanism of pathogenicity. Microbiol Rev 1982;46:162-70. 2. Frach CE. Status of a group-B Neisseria meningitidis vaccine. Eur J Clin Med 1985;4:504-10. 3. Guillén G, Silva R, Álvarez A, Coizeau E, Novoa L, Selman M, et al. Cloning and expression in E. coli of a high molecular weight outer membrane protein (PM6). In: Conde-Glez CJ, Morse S, Rice P, Sparling F, Calderon E, editors. Pathobiology and Immunology of Neisseriaceae. Proceeding of the 8th International Pathogenic Neisseria Conference; 1992 Oct 4-9; Cuernavaca, Mexico . Cuernavaca: INSP; 1994. p.834-840. 4. Silva R, Selman M, Guillén G, Herrera LS, Fernández JR, Novoa LI, et al, inventors; CIGB, assignee. Nucleotide sequence Neisseria meningitidis, its use in vaccine preparations. Europ Pat Aplication EP 0474313 A2. 1992. 5. Bringas R, Fernández J. A lipoamide dehydrogenase from Neisseria meningitidis has a lipoyl domain. Protein Struct Func Genet 1995;21:303-6. 6. Reed RJ. Multienzyme complexes. Acct Chem Res 1974;7:40-6 7. Mattevi A, de Kok A, Perham RM. The pyruvate dehydrogenase multienzyme complex. Curr Opin Struct Biol 1992; 2:877-87. 8. Perham RN. Domains, motif and linkers in 2-oxo-acid dehydrogenasa multienzyme complexes: a paradigm in the design of a multifunctional protein. Biochem 1991;30:8501-12. 9. Smith LD, Burgard SJ, Danson MJ, Hough DW. Dihydrolipoamide dehydrogenase from the thermo acidophilic archaebacterium Thermoplasma acidophilum. Biochem Soc Trans 1987;15: 1097. 10. Danson MJ, Conroy K, McQuattie A, Stevenson KJ. Dihydrolipoamide dehydrogenase from Trypanosoma brucei. Characterization and celular location. Biochem 1987;243:661-5. 11. Owen P, Kaback HR, Graeme-Cook KA. Identification of antigen 19/27 as dihydrolipoyl dehydrogenase and its probable involvement in ubiquinone-mediated NADH-dependent transport phenomena in membrane vesicles of Escherichia coli. FEMS Microbiol Lett 1980;7:345-8. 12. Reed LJ, Hackert ML. Structure-Function relationships in dihydrolipoamide acyltranferases. J Biol Chem 1990; 265: 8971-4. 13. Hein S , Steinbuchel A. Biochemical and molecular characterization of the Alcaligenes eutrophus pyruvate dehydrogenase complex and identification of a new type of dihydrolipoamide dehydrogenase. J Bacteriol 1994;176: 4394-4408. 14. Kruger N, Oppermann FB, Lorenz H, Steinbuchel A. Biochemical and molecular characterization of the Clostridium magnum acetoin dehydrogenase enzyme system. J Bacteriol 1994;176:3614-30. 15. Zhu PP, Peterkofski A. Sequence and organization of genes encoding enzymes involved in pyruvate metabolism in Mycoplasma capricolum. Protein Sci 1996;5: 1719-36. 16. Massey V. The identity of diaphorase and lipoyl dehydrogenase. Biochim Biophys Acta 1960;37:314-22. 17. Massey V, Sandro G. Role of charge-transfer interactions in flavoprotein catalysis. Ann Acad Sci New York 1974;227: 446-65. 18. Reed LJ, Koike M, Levich ME, Leach FR. Studies on the nature and reactions of protein-bound lipoic acid. J Biol Chem 1958;232:143-58. 19. Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248-54. 20. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227:680-5. 21. Yau WW, Kirkland JJ, Bly DD. Modern size-exclusion liquid chromatography. New York: John Wiley & Sons Inc.; 1979. 22. De la Sierra I, Pernot L, Prangé T, Saludjian P, Schiltz M, Furme R, et al. Molecular Structure of the Lipoamide Dehydrogenase Domain of a Surface Antigen from Neisseria meningitidis. J Mol Biol 1997;269:129-41. 23. Hemmerich P, Nagelshneider G, Veeger C. Chemistry and biology of flavins and flavoproteins. FEBS Lett 1970;8:69-83. 24. Williams CH. Flavin-containing dehydrogenase. The enzymes. 3rd ed. New York: Acad New York; 1976. Recibido en julio de 1998. Aprobado en septiembre de 1998. Copyright 1999 Elfos Scientiae The following images related to this document are available:Line drawing images[ba99014a.gif] [ba99014d.gif] [ba99014b.gif] [ba99014e.gif] [ba99014c.gif] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}