 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Biotecnología Aplicada 1999;16:109-111 Presencia de la mutación G1138A del gen del R3FCF en un grupo de pacientes acondroplásicos cubanos @ Thelvia I Ramos Gómez, Estela Morales Peralta,
Teresa Collazo Mesa, Centro Nacional de Genética Médica. Centro
colaborador de la OMS para el desarrollo de enfoques
genéticos en la promoción de la salud. Instituto
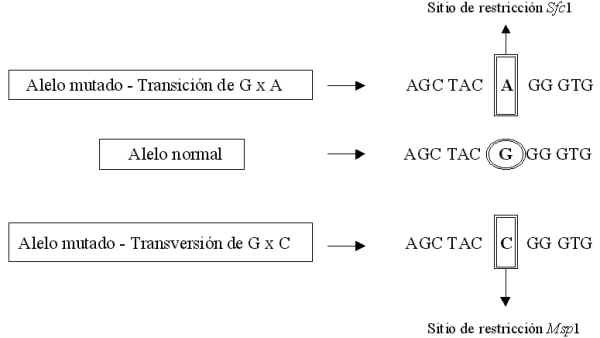
Superior de Ciencias Médicas de La Habana, Cuba. Code Number:BA99019 ABSTRACT Achondroplasia, inherited as an autosomal dominant trait, is the most common human skeletal dysplasia. Prevalence at birth in Cuba was estimated in 1/25000. Achondroplasia locus was assigned to chromosome 4p16.3 and includes the coding region for fibroblast growth factor receptor 3 (FGFR3). Two point mutations in exon 10 of the FGFR3 gene (G1138A and G1138C) have been described. These mutations create new restriction sites for Sfc1 (the most frequent mutation) and Msp1. Genomic DNA isolated from blood samples of 40 Cuban patients (24 relatives and 16 sporadic cases) was studied. A region of 164 bp that includes the transmembrane domain of FGFR3 was amplified by PCR and the amplicons were analized with the restriction enzimes Sfc1 and Msp1. All affected individuals showed fragment sizes of 109 and 55 bp corresponding to the digestion by Sfc1; therefore, they had the same mutation G1138A. Keywords: achondroplasia, autosomal dominant, FGFR3, hipoachondroplasia, osteochondrodysplasias, thanatophoric dysplasia RESUMEN La acondroplasia, de herencia autosómica dominante, es la más común de las displasias esqueléticas humanas. En Cuba, su prevalencia en los nacidos ha sido estimada en 1/25 000. El locus de la acondroplasia fue asignado al cromosoma 4p16.3 y codifica el receptor 3 del factor de crecimiento fibroblástico (R3FCF). Se han descrito dos mutaciones puntuales en el exón 10 del gen del R3FCF (G1138A y G1138C). Estas mutaciones crean nuevos sitios de restricción para las enzimas Sfc1 (la mutación más frecuente) y Msp1. Se estudió el ADN genómico aislado a partir de 40 muestras de sangre de estos pacientes cubanos (24 familiares y 16 esporádicos). Mediante PCR, se amplificó una región de 164 pb que incluye el dominio de transmembrana del R3FCF y el producto de la reacción se analizó con las enzimas de restricción Sfc1 y Msp1. Todos los individuos afectados mostraron fragmentos de talla de 109 y 55 pb, correspondientes a la digestión con Sfc1. Por lo tanto, ellos tenían la misma mutación G1138A. Palabras claves:acondroplasia, autosómica dominante, displasia tanatofórica, hipoacondroplasia, osteocondrodisplasias , R3FCF Introducción La acondroplasia (MIM 100800) es una afección autosómica dominante considerada como el enanismo de miembros cortos más frecuente entre los seres humanos. Su frecuencia ha sido estimada entre 1/15,000 y 1/77,000 [1-3]. De 80-90% de los casos son esporádicos, lo que ha sido asociado con una avanzada edad paterna; el resto es hereditario [1,3-5]. En nuestro país, basado en los datos obtenidos a través del Registro Cubano de Malformaciones Congénitas (RECUMAC), se calculó su prevalencia en los nacidos en 1/25,000 (Alonso F, 1997). En la literatura cubana no existen otros reportes científicos relacionados con esta patología. En la mayoría de los pacientes, el genotipo se corresponde con su estado heterocigótico, sin embargo, se han descrito casos de hijos homocigóticos de padres acondroplásicos heterocigóticos como una forma letal de la enfermedad, trayendo consigo el fallecimiento del individuo generalmente antes del primer año de vida [5,6]. La historia natural de la enfermedad se caracteriza porque los pacientes, además de una baja talla desproporcionada evidente al nacer, manifiestan complicaciones que afectan su calidad de vida. Varios autores han detallado sus aspectos clínicos y sus estudios radiológicos [7]. El gen de la acondroplasia ha sido ubicado en el cromosoma 4p16.3, confinado a una región candidata de 2,5 megabases que se extiende desde el locus D4S43 hasta el telómero [8]. Más tarde, Shiang y colaboradores identificaron mutaciones específicas para esta displasia esquelética en el gen que corresponde al receptor 3 del factor de crecimiento fibroblástico (R3FCF). Posteriormente, se identificaron mutaciones puntuales en el exón 10 de este gen, las cuales consisten más frecuentemente en una transición de G por A en el nucleótido 1138, y en una menor proporción, en una transversión de G por C en el mismo sitio. Debido a la degeneración del código genético, ambas mutaciones dan lugar a una sustitución de glicina por arginina en el codón 380, que corresponde al dominio de transmembrana del R3FCF [9] (Figura). Figura. Mutaciones en el exón 10 del gen del R3FCF, correspondiente al dominio de transmembrana. Al ampliarse el número de acondroplásicos estudiados, se observó un alto grado de homogeneidad genética, pues se encontraba una de esas dos mutaciones en la mayoría de los casos [10-12]. Sin embargo, Superti-Furga y colaboradores, en 1995, describieron el caso de un recién nacido acondroplásico que no tenía ninguna de las dos mutaciones del nucleótido 1138, pero que presentaba una sustitución de glicina por cisteína en el codón 375 del gen del R3FCF [13], lo que indica heterogeneidad alélica para este gen y, además, reafirma el papel etiológico del R3FCF en la patogénesis de la acondroplasia. Utilizando el método descrito por Shiang y colaboradores [9], que permite detectar las mutaciones que causan un cambio de glicina por arginina en el codón 380 del gen del R3FCF, se decidió estudiar un grupo de pacientes cubanos heterocigóticos para la acondroplasia con el propócito de conocer el comportamiento de estas mutaciones en nuestro medio y mejorar el diagnóstico y el asesoramiento genético que se le ofrece a estos pacientes. Materiales y Métodos Se estudiaron 40 pacientes cubanos de diagnóstico confirmado a través del cuadro clínico y/o radiológico, y se completó la investigación familiar, lo que permitió la confección de los árboles genealógicos. El estudio molecular se realizó después de que cada paciente dio su consentimiento; en caso de ser menor de edad, se solicitó la autorización de los padres o tutores. Extracción de ADN Se extrajo el ADN a partir de los leucocitos obtenidos de 10 mL de sangre periférica, con la utilización de EDTA 56 mg/mL como anticoagulante. El ADN así obtenido se purificó por el método de precipitación salina [14]. Amplificación por PCR Se utilizó, en principio, el método descrito por Shiang y colaboradores [9] mediante la reacción en cadena de la polimerasa (PCR) a partir de ADN genómico. Las condiciones empleadas fueron las siguientes: 100 ng de ADN, 2U de Taq ADN polimerasa por reacción, 25 pmol de cada cebador, dNTP 2 µM, MgCl2 10µM en un volumen de reacción de 40 µL. Se utilizaron los oligonucleótidos: 5'-AGG AGC TGG TGG AGG CTG A-3¢ (oligo A) y 5'-GGA GAT CTT GTG CAC GGT GG-3' (oligo B). La reacción de amplificación consistió en 5 min a 94°C seguido de 35 ciclos de desnaturalización (30 s a 94 °C), hibridación de los cebadores (30 s a 65°C) y extensión (30 s a 72 C). Al finalizar, se realizó una extensión durante 5 min a 72 C. La reacción se efectuó en un termociclador modelo Mastercycler 5330. Análisis de restricción El producto del PCR fue digerido con Sfc1 y/o Msp1, empleándose 40µL del PCR y 4U de cada enzima, durante 1-2 h a 37 C. Se realizó una electroforesis del producto de la digestión en un gel de agarosa a 2%. Las bandas se visualizaron mediante tinción con bromuro de etidio. Resultados y Discusión Los factores de crecimiento fibroblástico son miembros de una familia de polipéptidos que son potentes reguladores del funcionamiento, la diferenciación y la proliferación celular. Los receptores de los factores de crecimiento fibroblástico tienen su expresión durante la embriogénesis, lo que sugiere que median diferentes funciones de los FCF durante el desarrollo [15]. La acondroplasia se debe a una alteración del R3FCF [9, 12, 16], y el sitio donde se encuentra ubicado este gen (cromosoma 4p16.3) aparece relacionado con varias osteocondrodisplasias. Los primeros estudios encontraron puntos de ruptura cromosómica sobre 4p16.3 en afecciones como la displasia tanatofórica y el síndrome polidactilia costillas cortas (una condrodisplasia autosómica recesiva) [17]. Posteriormente, se reportó que el gen del R3FCF interviene en la patogénesis de otras displasias esqueléticas como la hipoacondroplasia, la displasia tanatofórica y la propia acondroplasia [18-21]. Estas enfermedades varían en cuanto a su cuadro clínico y a su severidad, por lo que la hipoacondroplasia (una de las formas alélicas de este gen) es mucho más leve que la displasia tanatofórica; esta última es letal al nacimiento. Con todos estos argumentos, se ha sugerido que el brazo corto del cromosoma 4 puede contener varios genes determinantes o responsables de las osteocondrodisplasias, o que estas enfermedades resultan de diferentes mutaciones de un mismo gen [22]. Francomano y colaboradores plantearon que en la acondroplasia la mutación ocurre en el cromosoma paterno, y sugirieron la influencia de factores en la replicación del ADN o en la reparación del mismo durante la espermatogénesis, lo que puede predisponer a la ocurrencia de la enfermedad [23]. Al caracterizar molecularmente los pacientes con esta afección, se ha observado que 97% presenta una transición de G por A, lo que constituye la mutación más común [9, 11]. En nuestro estudio, todos los pacientes presentaron la misma mutación, resultado que correspondió con la mayoría de los informes previos: Shiang R (1994) [9]; Rosseau F (1994) [11]; Bellus GA (1995) [12]; Stoilov L (1995) [24]; Kaysereli H (1996) [25]. De los 40 pacientes cubanos estudiados, 24 eran familiares y 16 casos esporádicos. La distribución según el color de la piel mostró 52,5% de individuos blancos y 47,5% de negros y mestizos. En la caracterización molecular, se concluyó que todos los acondroplásicos estudiados presentaron la mutación más frecuente, es decir, que en el nucleótido 1138 existía una transición que resulta en un cambio de glicina por arginina (Tabla). Esta mutación es detectada por digestión con la enzima Sfc1, que rinde los siguientes fragmentos: uno de 164 pb correspondientes al alelo normal, y otros dos de 109 y 55 pb correspondiente al alelo mutado. No se obtuvo producto de la digestión con la enzima Msp1. Tabla. Mutaciones en la región del gen del FGFR3 correspondiente al dominio de transmembrana, en pacientes con acondroplasia. Se observa un mayor número de casos familiares con respecto a los esporádicos, debido a que en una de las familias estudiadas, 10 de sus miembros son acondroplásicos.
Es posible que cuando se investigue un mayor número de casos cubanos, se identifique en alguno la segunda mutación (cambio de G por C) u otra, debido a que según algunos autores, el gen del R3FCF es muy grande y, por lo tanto, pudieran existir otras mutaciones [12]. De hecho, Superti-Furga encontró una nueva mutación en el codón 375 de este gen, que resulta en una sustitución de glicina por cisteína [13]. Las otras posibles mutaciones pudieran llevar a fenotipos silentes y de ahí que aún no se hayan detectado. En los acondroplásicos estudiados se ha observado una gran homogeneidad genética, a pesar del elevado por ciento de mutaciones nuevas. Estas mutaciones aparecen en el contexto del dinucleótido CpG, el cual es conocido como hotspot (sitio caliente) para las mutaciones de transición, especialmente si el residuo de citosina está metilado [26, 27]. La desaminación de la 5-metilcitosina resulta en un residuo de timina, y cambia el par de bases G:C por el par T:A después de la replicación del ADN. Esta mutación es la más común en los acondroplásicos con cromosomas afectados que se han estudiado. Este sitio, en particular, parece tener un por ciento extraordinariamente alto de mutaciones, considerándose el nucleótido 1138 del gen del R3FCF, como el nucleótido más mutable en el genoma humano [16]. Parece que esta zona se corresponde con un sitio lábil del genoma, dada la alta tasa de mutaciones nuevas que tiene este gen, siendo la mayoría de los casos esporádicos. Conclusiones Todos los pacientes presentan la mutación principal, cambio de G por A en el nucleótido 1138, con una gran homogeneidad mutacional. Agradecimientos Expresamos nuestra gratitud a todos los pacientes por la confianza depositada en nosotros. Agradecemos al técnico Lázaro Antúnez por su valiosa colaboración. Referencias 1. Murdoch JL, Walker BA, Hall JG, Abbey H, Smith KK, Mckusick VA. Achondroplasia: a genetic and statistical survey. Ann Hum Genet 1970;33:227-4. 2. Gadner RJM. A new estimate of the achondroplasia mutation rate. Clin Genet 1977;11:31-8. 3. Oberklaid F, Danks DM, Jensen F, Stace L, Rosshandler S. Achondroplasia and hypoachondroplasia: comments on frequency, mutation rate and radiological features in skull and spine. J Med Genet 1979;16:140-6. 4. Stoll C, Dott B, Roth MP, Alembik Y. Birth prevalence rates of skeletal dysplasias. Clin Genet 1989;35:88-92. 5. Gorlin RJ, Cotten M, Levin LS. Syndromes of the Head and Neck. 3rd ed. New York: Oxford University Press; 1990.p.171-9. 6. Mckusick VA. Mendelian inheritance in man. Catalogues of autosomal dominant, autosomal recessive and X-linked phenotypes. The Johns Hopkins University Press. OMIN; 1996. 7. Nicoletti B, Kopits SE, Ascani E, Mckusick VA, editors. Human achondroplasia. A multidisciplinary approach. Proceedings of the First Symposium on Human Achondroplasia; 1986 Nov 19-21; Rome, Italy. New York: Plenum Press; 1988. 8. Francomano CA, Ortiz de Luna RI, Hefferon TW, Bellus GA, Turner CE, Taylor E, et al. Localization of the achondroplasia gene to distal 2,5 Mb of human chromosome 4p. Hum Mol Genet 1994;3:787-92. 9. Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, et al. Mutations in transmembrane domain of R3FCF cause the most common genetic form of dwarfism, achondroplasia. Cell 1994;78:335-42. 10. Velinov M, Slaugenhaupt SA, Stoilov I, Scott CI, Gusella JF, Tsipouras P. The gene for achondroplasia maps to the telomeric region of chromosome 4p. Nat Genet 1994;6:312-7. 11. Rousseau F, Bonaventure J, Legeall-Mallet L, Pelet A, Rozet JM, Maroteaux P, et al. Mutations in the gene encoding fibroblast growth factor receptor in achondroplasia. Nature 1994;371: 252-4. 12. Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, et al. Achondroplasia is defined by recurrent G380R mutations of R3FCF. Am J Hum Genet 1995;56:368-73. 13. Superty-Furga A, Eich G, Bucher HU, Wisser J, Giedion A, Gitzelmann R, et al. A glycine 375 to cysteine substitution in the transmembrane domain of fibroblast growth factor receptors in a newborn with achondroplasia. Eur J Pediat 1995;154: 215-9. 14. Miller SA. A simple salting out procedure for extracting DNA from human nucleoted cells. Nucl Acid Res 1988;16:1215-8. 15. R & D Systems-Catalogue. 1st ed. New York: Wiley-Liss;1995. p.123-6. 16. Chellaiah AT, McEwen DG, Werner S, Xu J, Ortiz DM. Fibroblast growth factor receptor 3 (R3FCF). J Biol Chem 1994;269:11620-7. 17. Rivas F. Chromosome 4 and patient with lethal short rib-polydactyly syndrome. Clin Genet 1987;31:97-101. 18. LeMerrer M, Rousseau F, Legeai-Mallet L, Landais JC, Pelet A, Bonaventure J, et al. A gene for achondroplasia-hipoachondroplasia maps to chromosome 4p. Nat Genet 1994; 6:314-7. 19. Bellus GA, McInstosh I, Smith EA, Aylsworth AS, Katila I, Horton WA, et al. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hipoachondroplasia. Nat Genet 1995;10:357-9. 20. Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS, et al. Thanatophoric dysplasia (types 1 and 2) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet 1995;9:321. 21. Rousseau F. Stop codon R3FCF mutations in thanatophoric dwarfism type 1. Nat Genet 1995;10:11-2. 22. Urioste M. Chromosome 4p and osteochondroplasias. Nat Genet
1994; 23. Szabo J, Bellus GA, Kaitila I, Francomano CA. Fibroblast growth factor receptor 3 (R3FCF) mutations in sporadic cases of achondroplasia occur exclusively on paternally-derived chromosome [abstract]. Am J Hum Genet 1996;59(4):A287. 24. Stoilov I, Kilpatrick MW, Tsipouras P. A common R3FCF gene mutation is present in achondroplasia but not in hipoachondroplasia. Am J Med Genet 1995;55:127-33. 25. Kayserill H. Prenatal diagnosis of achondroplasia by direct sequence analysis of the transmembrane domain of R3FCF gene [abstract]. Am J Hum Genet 1996; 59(4):A323. 26. Steinberg RA, Gorman KB. Linked spontaneous CG-TA. Mutations at cp. sites in the gene for protein kinase regulatory subunit. Mol Cell Biol 1997;12:767-72. 27. Barker D, Schafer M, White R. Restriction sites containing pCpG show a higher frequency of polymorphisms in human DNA. Cell 1992;36:131-138. Recibido en julio de 1998. Aprobado en diciembre de 1998. Copyright 1999 Elfos Scientiae The following images related to this document are available:Photo images[ba99019a.jpg] | |||||||||||||||||
| |||||||||

{kind=link}