|
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
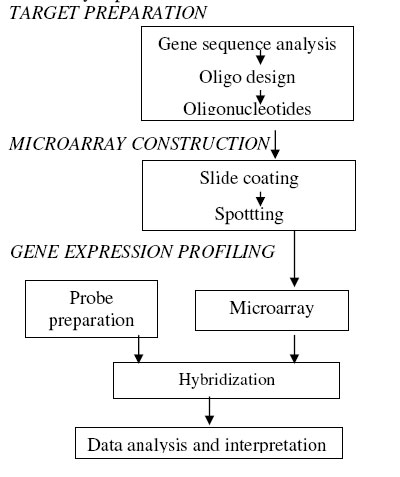
Biokemistri, Vol. 18, No. 1, June, 2006, pp. 1-7 Systems Biology and the Development of Vaccines and Drugs for Malaria Treatments Ezekiel F. ADEBIYI Department Computer and Information Sciences, College of Science and Technology, Covenant University, P.M.B 1023, Ota, Nigeria E-mail: eadebiyi@sdsc.edu;Tel: 01-7924130 Received 16 November 2004 Code Number: bk06001 Abstract The sequencing race has ended and the functional race has already begun. Microarray technology enables simultaneous gene expression analysis of thousands of genes, enabling a snapshot of an organisms’ transcriptome at an unprecedented resolution. The close correlation between gene transcription and function, allow the inference of biological processes from the assessed transcriptome profile. Among the sophisticated analytical problems in microarray technology at the front and back ends respectively, are the selection of optimal DNA oligos and computational analysis of the genes expression. In this review paper, we analyse important methods in use today in customized oligos design. In the course of executing this, we discovered that the oligos designer algorithm hanged on gene PFA0135w of chromosome 1, while designing oligos for the gene sequences of Plasmodium falciparum. We do not know the reason for this yet, as the algorithm runs on other sequences like the yeast (Saccharomyces cervisiae) and Neurospora crassa. We conclude the paper highlighting the procedures encompassing the back end phase and discuss their application to the development of vaccines and drugs for malaria treatment. Note that, malaria is the cause of significant global morbidity and mortality with 300-500 million cases annually. Our aims are not ends, but a means to achieve the following: Iterate the need for experimental biologists to (i) know how to design their customized oligos and (ii) have some idea about gene expression analysis and the need for cooperation between experimental biologists and their counterpart, the computational biologists. These will help experimental biologists to coordinate very well the front and the back ends of the system biology analysis of the whole genome effectively. Key words: bioinformatics, vaccines, drugs, malaria. Introduction The expression "systems biology" is often used today to describe attempts at unraveling molecular systems (the function of the genome), beyond the traditional level of single genes and single proteins, focusing on the intricate circuitry that governs growth, development, homeostasis, behaviour and the onset of diseases, which is largely controlled by the RNA and proteins encoded by the cognate genes and the complex and dynamic interaction of the genes with the environment. A detailed conceptual view of gene regulatory circuitry in organisms will require extensive expression monitoring at the level of the whole genome. The challenge of this biological analysis requires the development and implementation of sophisticated analytical methods. DNA microarray technology offers a great tool for these tasks10. The basic steps to study the function of a whole genome using system biology include the following6: (i) identify all the players of the system, that is, all the components that are involved (e.g. genes, proteins, compartments), (ii) perturb each component through a series of genetic or environmental manipulations and record the global response using high-throughput technologies (e.g. microarrays), and (iii) build a global model and generate new testable hypotheses. Return to (ii) and in some cases to (i) when missing components are discovered. In microarray term, these steps are (i) design of the DNA chips (arrays), (ii) gene expression profiling experiments, and iii) computational analysis of the array data3. DNA chips are glass surfaces bearing thousands of DNA fragments at discrete sites at which the fragments are available for hybridization. Hybridization of fluorescently labelled RNA and DNA-derived samples to DNA chips allows the monitoring of gene expression or occurrence of polymorphisms in genomic DNA17. Two DNA chip formats currently in wide use are the cDNA array format and high density synthetic oligonucleotide array format. Oligonucleotide expression arrays include both short oligo (20-25 mers) arrays (Affymetrix geneChip) and long oligo (50-70 mers) arrays. It has been noted in Le Roch et al.11 that longer oligos than 25-mer (that could be hybridization problematic) might be needed to generate accurate expression profiles. Nevertheless, Affymetrix has combined oligonucleotide synthesis and photolithographic computer chip synthesis to generate DNA chips that display 40 000-65 000 DNA short oligonucleotides which represent up to 9000 genes on a 1.6 cm2 glass surface. Here, we focus on the design of the DNA chips (the high density synthetic oligonucleotide array format) and the computational analysis of the array data. Microarray experiment The following figure shows the basic procedures involved in the set up of a microarray experiment.
A microarray experiment based on an in-laboratory custom built auxillaries is less expensive and made possible via many available free software and protocols. Microarrays can be made from either the spotted glass slide or the Affymetrix GeneChip. Affymetrix chips are high density array. The technology involved is highly standardized and are exclusively provided by Affymetrix. Hence the chips and the required equipments are expensive. Spotted glass slide microarrays made from microscope glass slide are significantly cheap and can be produced by any laboratory equipped with the necessary instruments for DNA production, purification, analysis and spotting. Functional information about the genes of an organism represented on the microarrays via oligonucleotides (oligos) are measured by their hybridization density with the probe. The probe here represents the RNA at all or some of its life cycle. In data analysis and interpretation, it is popular to use the one-way ANOVA statistical test to identify differentially expressed genes from the time course data19 and cluster analysis to classify genes to be of similar functions based on their similar expression profiles4. A recent work of statistical analysis on gene expression can be found in Wichert et al18, while the genes network3,9 (also on the computational side), an alternative to cluster analysis, provides another strategy to elucidate gene functions. Selection of Optimal DNA Oligos for gene expression arrays This was done at the target preparation step of a microarray experiment. How should the oligos be chosen? One thing is clear; each oligo should be specific for exactly one gene. This is because, in the experiment, only the fluorescently labelled transcripts of a single gene should hybridize to a given oligo, so the measurement taken at the oligos spot can be interpreted as the corresponding genes expression level in the sample. Note that the cDNA or mRNA from the sample need not contain the perfect Watson-Crick complement of an oligo to hybridize. Computationally, using the terminologies from Rahmann (2002)15, given two sequences, the master sequence, the DNA sequence for which we wish to design oligos (usually some or the whole part of the coding region of a genome, in other words, a list of genes) and the background sequence (usually consists of all known genes and/or their reversed complements), the oligos design problem is to select patterns of size L (this is usually peck at 25, 50 , or 70) at more or less constant melting temperature (usually denoted as TM) that are specific (unique) to each gene (note that more than one oligos may represent a gene), allowing very small percentage of similarity to the background sequence at m-mismatches. The melting temperature TM of an oligo-cDNA duplex is the temperature at which half of the potential duplexes have hybridized and are in the DNA-typical double-helical state, and the other half is still single-stranded. Note that, in principle, the melting temperature of perfect as well as approximate Watson-Crick paired oligos, can be computed assuming a simple nearest-neighbour model16. Common additional parameters include the GC content of the oligo, its distance from the 3' end of the background sequence, and minimum distance between the 5' ends of two adjacent oligos, if two or more oligos are representing a gene. Furthermore, sequences that are internal self binding (form secondary structure), of low-complexity and contain 'GGGGG', 'CCCCC', 'TTTTT', or 'AAAAA' are avoided in the oligos. For example, we used the algorithm of Bozdech et al.3 to identify the oligos of length 70 (one per gene) with GC content equal 28 from the Plasmodium falciparum. Their results for falcipain 2, a gene involved in chloroquine resistance are as follows: 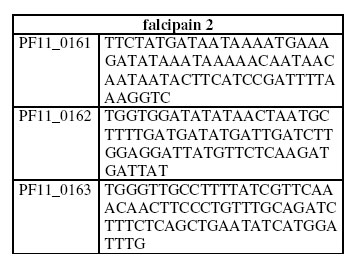 A straightforward (but lazy) approach is to consider as candidate oligos, all possible L-mers contained in the master sequence, filtering out all L-mers that are repeats. We have developed tool for this in Adebiyi et al1. Other filtering parameters can then follow as discussed above. Existing oligos selector algorithms include that of Kaderali and Schliep7, Li and Stormo13, Rouillard, Herbert and Zuker14 and Rahamann15. Other algorithms include Lockhart et al.10 and Bozdech et al.2. A computational analysis of the methods use to select oligos is important since the correctness of the front-end, of course has great influence on the results we will obtain at the middle and back ends respectively. Our computational analysis is built using suffix tree8 with the algorithms described by Gusfield5. Performing tasks to computationally analyze important methods in use today to design customized oligos, the algorithm of Rouillard et al.14 hanged on gene PFA0135w of chromosome 1, while designing oligos for publicly available genome of Plasmodim falciparum (see acknowledgement). Their algorithm does not behave this way when we used it on Yeast (Saccharomyces cervisiae) and Neurospora crassa genes. We further discovered that the results of the four algorithms (Li and Stormo13, Rouillard et al.14, Rahamann15 and Bozdech et al.2) tested, are at variance. This poses serious hindrance to the development of microarrays using the oligonucleotides approach. And since this approach has significant advantage over its alternative13, our finding thus calls for further work in this area. A further work on this problem and detail comparative analysis of existing methods are presently been undertaken by us. Development of vaccines and drugs for malaria treatments After the completion of the genome sequence for Plasmodium falciparum, Le Roch et al.12 carried out a large scale microarray experiment to determine the functions (of more than 95%) of the predicted Plasmodium falciparum genes as the parasite moves through its life cycle, using an Affymetrix chip containing 260,596 25-nucleotide single-stranded probes from predicted coding sequence (5159 genes) that include mitochondrion and plasmid sequences. This study12 is important because the spread of malaria relies on finely tuned reciprocal interactions between Plasmodium falciparum and the Anopheles mosquito vector, which allow the parasite to complete its cycle without seriously affecting the vectors’ fitness. Therefore, a comprehensive understanding of the delicate interactions between Anopheles and Plasmodium can serve the development of malaria control strategies based on blocking the parasite’s development in the mosquito. Le Roch et al.12,13 examined nine different stages of the parasite development, namely mosquito salivary gland sporozoites, which infect humans; seven periodic erthrocytic asexual stages and lastly, the sexual stage gametocyte, the form by which the parasite is transmitted from humans to mosquitoes. It was discovered that 4557 genes (88% of the predicted genes) were expressed in at least one stage of the life cycle. A total of 602 genes were not expressed in any stage examined, and a majority (87%) of this code for hypothetical proteins that may be expressed at other stages of the life cycle. In a personal communication, Le Roch confirmed that few of these genes may be expressed at a background level, thus making them difficult to detect, but a guess is that most of them should be expressed during the liver stages or/and mosquitos gut stages. In a further analysis, Le Roch et al. (2003)12 discovered that out of the 4557 genes that expressed themselves, 2235 genes were expressed at some stages, while the remaining 2322 genes were designated as constitutively expressed, since they are expressed at all examined stages. They noted that, this group contains genes coding for both uncharacterised hypothetical proteins and house keeping proteins and many of the hypothetical proteins are likely involved in maintenance of the parasite function throughout the life cycle, and those without human orthologs might represent targets for drug development. Cluster analysis provided 15 cluster groups. The genes in each group are of similar expression profiles, thus assumed to be of similar function. Each group also contains genes, whose functions are known. The highest probability obtained that any of these groups overlaped by chance with these known genes is 0.029. These clusters provided highly probable genes that may represent targets for functional disruption of the liver stage invasion process (either by small molecule interactions or vaccination), transmission blocking interventions, drugs focusing on disruption of the parasite at its replicating stage, etc. Notably among their results is that, cluster 7 contains plasmepsin, pfCRT, and falcipain 2, a chloroquine resistance transporter gene. Further work carried out in the laboratories of Dimoupoulos, DeRisi, Kissinger and Kafatos has been done to uncover more correctly and completely, the functions of the predicted genes of Plasmodium falciparum at some stages of its life cycle instead of considering a whole lot of its development stages as Le Roch et al. (2003)12 did. Furthermore, the Plasmodium parasites suffer large losses during their passage through the vector mosquito. The mechanisms underlying parasite elimination in the mosquito are unknown. Several lines of evidence have linked mosquito robust immune responses to the documented Plasmodium losses without clearly indicating the stage specificity and the killing mechanisms. Nor have the activation of specific immune genes been linked with specific elicitors of malaria infected blood. The further work listed above also dwelled on this challenge. Conclusion and Open Problems Malaria is one of the most serious diseases causing 1.5 to 2.7 million deaths every year, mostly among children in sub-saharan Africa, including Nigeria. The favourable climatic changes for the mosquitoes, the lack of effective vaccines, the development of parasite resistance to drugs and mosquito resistance to insecticides contribute to the expansion of the disease, and have created an acute need for the development of additional control strategies. None of the popular laboratories working on microarray experiment application to malaria treatment is located in Nigeria. Some are even located in WHO (World Health Organization) certified malaria free countries. In this paper, in an attempt to develop an optimal oligos selection algorithm, we analyzed existing methods used today in customized oligo design. In the course of doing this, we discovered that the set of oligos designed by each algorithm for the same gene sequences are at variance and OligoArray2.017 will not work on gene PFA0135w of chromosome 1 of the genome of P. falciparum. We also uncovered the following dry and wet laboratory open problems, namely: (i) development of better statistical analysis methods for gene expression18, (ii) the design of better computational methods to derive the genes network from gene expression data3, (iii) develop further dry/wet laboratory tools to streamdown / validate thousands of proteins identified in the P. falciparum genome as new drugs or vaccine targets. and (iv) Le Roch et al.12 noted that the genome sequence on their array were derived from an isolated, 3D7, that has been maintained in culture for generations, and the RNA samples were also derived from parasites that had been maintained in continuous culture for generations; therefore the obtained expression patterns may not reflect those that exist in parasites replicating human hosts. Obtaining the sequence of a wild P. falciparum isolate and obtaining expression profiles of parasites isolated directly from humans will improve the biological relevance of the studies. This is an open problem for the Nigerian experimental biologists. Acknowlegement We thank Prof. Stormo and Dr. Rouillard for sending useful programs. Sequence data for P. falciparum was obtained from The Sanger Institute website. REFERENCES
© 2006 Nigerian Society for Experimental Biology. |
| |||||||||