 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
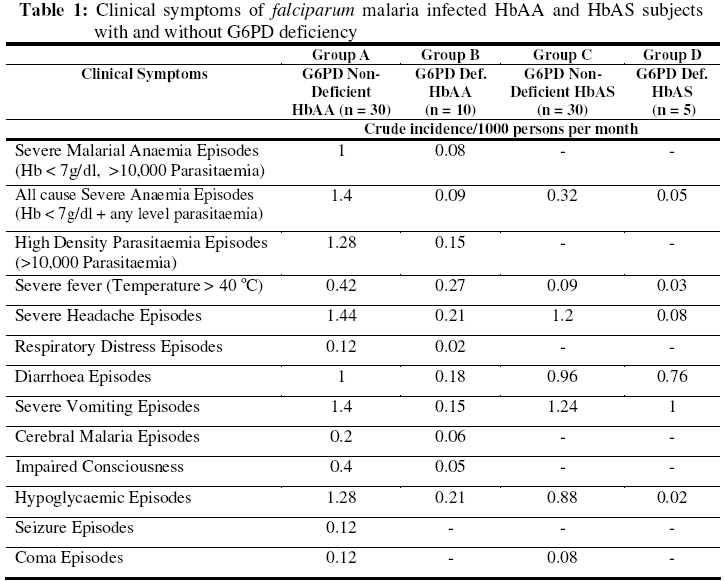
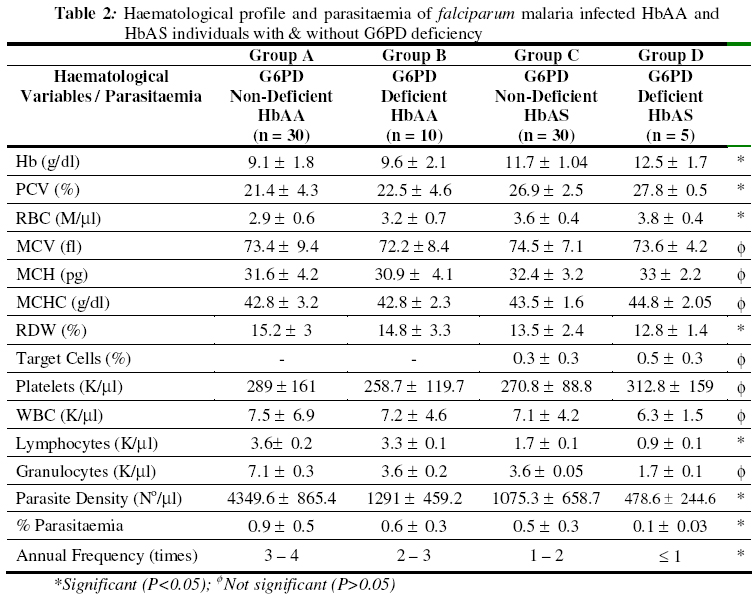
Biokemistri, Vol. 18, No. 2, December, 2006, pp. 89-97 Influence of sickle heterozygous status and glucose-6-phosphate dehydrogenase deficiency on the clinico-haematolgoical profile of Plasmodium falciparum-infected children Francis M. AWAH* and Pete N. UZOEGWU Department of Biochemistry, University of Nigeria, Nsukka, Nigeria *Author to whom correspondence should be addressed. E-mail: awambuh@yahoo.com, Tel: +234-803-4418969 Received 22 September 2006 Code Number: bk06014 Abstract Sickle haemoglobin (HbS) and glucose–6–phosphate dehydrogenase (G6PD) enzyme deficiency genes are known to offer reliable protection against falciparum malaria in malaria endemic areas of the world. However, the mechanism of protection is not yet completely understood. In this study, we investigated the contribution of HbS and G6PD enzyme deficiency status in ameliorating the severity of malaria attack by comparing the clinical symptoms, parasitaemia and haematological profiles of Plasmodium falciparum–infected volunteer children. The selected group of children, G6PD deficient sickle heterozygotes (HbAS) (n = 5), G6PD non–deficient HbAS (n = 30), G6PD deficient dominant homozygotes (HbAA) (n = 10) and G6PD non–deficient HbAA (n = 30) were monitored for a period of one year with a view to elucidating further the involvement of HbS and G6PD enzyme deficiency in the protection of children against plasmodial infection. Results revealed greater severity (indicated by malarial anaemia), higher incidence of atypical thrombocytopenia, high white blood cell (WBC) counts and significantly higher (P < 0.05) parasite density and percentage parasitaemia in G6PD non–deficient HbAA subjects compared to G6PD non–deficient HbAS, G6PD deficient HbAS subject and G6PD deficient HbAA. Less severe clinical malarial symptoms were also observed more in G6PD deficient HbAS when compared to G6PD non-deficient HbAA subjects during malaria attack. These results seem to indicate that inheriting both genetic defects reduces the profligacy of malaria parasite and hence, ameliorate the severity of acute falciparum malaria. Consequently, selective advantage against fatal falciparum malaria seems to be conferred since malarial anaemia, parasitaemia and severe malarial symptoms were significantly reduced. Keywords:falciparum malaria, clinico–haematological profile, sickle haemoglobin, G6PD deficiency INTRODUCTION Plasmodium falciparum, a mosquito–borne haematoprotozoan parasite, is the main cause of disease and death in African countries1,2. As a result of heterozygous and hemizygous advantage against malaria, the inherited haemoglobinopathies and red cell enzymopathies are the commonest monogenic diseasesin the world3. Both sickle cell trait and G6PD deficiency co-exists in almost 11% American black population4, as well as in Cameroonian populations5. Glucose-6-phosphate dehydrogenase (G6PD) one of the most common X-linked recessive hereditary enzymopathy affecting erythrocyte metabolism6 catalyses the first step of the pentose phosphate pathway (PPP), leading to the production of reduced nicotinamide adenine dinucleotide phosphate (NADPH) within the erythrocytes and playing a critical role in the metabolism of glucose and in maintaining the balance of reduced/oxidized state of glutathione, an antioxidant in cells. Reduced glutathione (GSH) helps to maintain a reduced environment within the red cell and enhances the ability of the cell to withstand haemolysis that could result from oxidative damage following exposure to oxidant drugs such as primaquine7,8,9. Increased oxidative stress in G6PD deficient cells and haemolytic episodes related to altered PPP and glutathione metabolism is well documented7. Plasmodium infected G6PD deficient cells are thought to impair parasite growth, as they serve as an unstable host for the parasite or behave as a suicidal package being rapidly sequestered in the spleen once it is infected10. Sickle haemoglobin (HbS) gene is associated with a point mutation at the sixth codon of the β-globin gene resulting in the synthesis of mutant HbS (β6 Glu→Val). Although HbS homozygousity is associated with sickle cell anaemia (SCA), individuals heterozygous for HbS variant, are said to be protected against severe malaria11. Haematological crisis in SCA are manifested by a sudden exacerbation of anaemia with a drop in haemoglobin concentration, probably due to polymerisation and destruction of HbS and acute splenic sequestration of deoxygenated sickled red blood cells. Aplastic crisis may also result in an attempt to compensate for destruction. The host’s clinical presentation, progress to disease and sometimes death from malaria infection is influenced by the host’s age, immunity to malaria, genotype and pregnancy, and by the species, virulence, strain and perhaps the geographic origin of the parasite12. Anaemia in P. falciparum malaria could be caused by such factors as bone marrow suppression, red blood cells haemolysis and ineffective erythropoiesis. Anaemia occurs when the concentration of haemoglobin falls below what is normal for a person’s age, gender and environment, resulting in the reduction of the oxygen carrying capacity of the blood8. Severe anaemia occurs 1.42 – 5.66 million times annually and kills 190,000 – 974,000 children less than five (5) years old annually13. Malarial anaemia is typically normocytic and normochromic, with a notable absence of reticulocytes, although microcytosis and hypochromia may be present in areas with high frequencies of α- and β-thalassaemia trait and/or iron deficiency14. Malaria resistance associated with sickle cell trait and G6PD deficiency has served as a prime example of genetic selection for over a century11. Nevertheless, the mechanism of this resistance remains a subject of considerable debate. While it probably involves innate factors such as the reduced ability of Plasmodium falciparum parasite to grow and multiply in HbAS and G6PD deficient erythrocytes, recent findings suggest that it might also involve the accelerated acquisition of malaria specific immunity12. Reports on the effect of G6PD deficiency on patients with sickle cell trait have been controversial. Bernstein et al. [15] claimed that the combination was beneficial while Steinberg et al.16 found no beneficial or adverse relation in co–inheritance. In this study we investigate the contribution of sickle haemoglobin and G6PD deficiency gene in the clinico–haematological profile of falciparum malaria infected subjects with intent to throw more light into the role these genetic defects could play in the protection against malaria severity. MATERIALS AND METHODS Study area and subjects A total of 75 volunteers aged between 1 to 18 years, living in the malaria endemic Ndop plain of North West province of Cameroon, selected after the haemoglobin genotype and G6PD deficiency status of 934 individuals were determined, participated in the study. The volunteers sorting malaria treatment at the Ndop District Hospital were divided into four cohort groups based on their G6PD deficiency status and haemoglobin genotype thus; 30 G6PD non–deficient HbAS subjects (group A), 30 G6PD non–deficient HbAA (group B), 10 G6PD deficient HbAA (group C) and 5 G6PD deficient HbAS subjects (group D). Selection was based on the individual’s accessibility and willingness to take part in monitoring for the malaria infection for a period of one year. Blood collection and Tests Blood samples (5ml) were collected by venipuncture into sample tubes containing EDTA as anticoagulant and then rocked gently to mix. However, blood samples (0.1ml) for routine monthly malaria test were collected by thumb pricking using lancets. Subjects were monitored for the frequency of malarial infection, parasite density, percentage parasitaemia, haematological profile, blood sugar level, and other clinical malaria symptoms such as impaired consciousness, respiratory distress, high fever, severe headache, diarrhoea, vomiting, weakness and anaemia for a period of one year. Preparation of haemoglobin lysate and determination of haemoglobin genotype Uncoagulated blood was centrifuged at 3,000 x g for ten minutes to separate the red blood cells from the plasma. The upper layer was aspirated out while the sediment was washed three times by re-suspending in equal volumes of normal saline (0.85% w/v) and then re-centrifuged at the same speed. The packed cells were re-suspended in equal volume of normal saline. 20ml of the suspended cells was then mixed with 80ml of distilled water in a 1:4 dilution to lyse the cells. The solution was gently shaken for two minutes and then centrifuged at 3,000 x g for 20 minutes. The resulting supernatant, Hb lysate, was used for the genotype test while the precipitate was discarded. The Hb lysate was stored in the refrigerator (2-4 oC) until used within four days. Alternately, 20μl of uncoagulated whole blood was mixed with 60μl of distilled water and the resulting Hb lysate spotted directly for Hb genotype determination. Haemoglobin genotypes were determined by cellulose acetate membrane electrophoresis (CAME) of Evans17 as modified by Uzoegwu18. Malaria diagnosis A total of 934 children (430 males and 604 females) were tested for malaria parasite based on clinical malaria symptoms and confirmed by microscopic examination of Giemsa or Leishman stained thick and thin blood films. The presence of even one parasite per microscopic field indicated positivity for malaria parasite. Determinations of parasitaemia and parasite density Parasitaemia and parasite density were determined according to the method of Greenwood and Armstrong19 as described in Awah & Uzoegwu5. Determination of G6PD deficiency The activity of G6PD was determined by fluorescent spot test as described by Beutler et al.20. The reaction mixture contained glucose-6-phosplate (0.01M), NADP+ (0.01M), saponins (0.02M), phosphate buffer, pH 7.4 and distilled water. Fluorescence was produced due to the reduction of NADP+ to NADPH. This reaction is coupled with oxidation of glucose-6-phosphate to 6-phosphogluconolactone and catalysed by G6PD. Specimen with G6PD activity of <20% of normal (severe deficiency) do not fluoresce as the small amount of NADPH formed is reoxidised by glutathione present in the reagent. Presence of fluorescence indicated normal cells while weak fluorescence indicated slight deficiency. Full blood count Haematological indices [erythrocyte counts, white blood cell count (WBC), lymphocyte counts (LYM), granulocyte counts (GRA), mean cell volume (MCV), mean cell haemoglobin (MCH), mean cell haemoglobin concentration (MCHC), packed cell volume (PCV) or haematocrit (HCT), Hb concentration and platelet count (PLT)] were analysed using an automated system, CELL-DYN 1200 Hematology Analyser (Abbott, USA). Determination of serum fasting blood sugar Fasting blood sugar (FBS) was assayed with a one-touch glucose-monitoring meter (Life Scan Inc., USA) as directed in the manufacturer’s operation manual. RESULTS Clinical symptoms during severe malarial infection Results showed that on a monthly basis a greater number of G6PD non-deficient HbAA subjects exhibited more frequent and severe clinical malarial symptoms when compared with G6PD non-deficient sickle heterozygotes, G6PD deficient dominant homozygotes (HbAA) and G6PD deficient sickle heterozygotes (HbAS). Results further showed that G6PD non-deficient HbAS subjects and G6PD deficient HbAS subjects had significantly fewer (P < 0.05) episodes of severe malarial anaemia and higher parasite density (Hb < 7 g/dl and >10000 parasites/μl) (Table 1). Haematological profile during falciparum Infection The data presented on Table 2 show that G6PD non-deficient HbAA subjects suffered more malaria attack, had significantly (P < 0.05) higher parasite densities and higher percentage parasitaemia than the G6PD deficient dominant homozygotes, G6PD non-deficient sickle heterozygotes and G6PD deficient HbAS groups. The results also revealed that G6PD deficient sickle heterozygotes had significantly higher (P < 0.05) Hb concentration, PCV, RBC count, RDW, than the G6PD non-deficient sickle heterozygotes, G6PD deficient dominant homozygotes and G6PD non-deficient HbAA subjects. However, G6PD non-deficient HbAA subjects had higher WBC and lymphocyte counts than G6PD deficient homozygotes, though statistically non significant (P > 0.05). Also, G6PD deficient sickle heterozygotes showed significantly higher (P < 0.01) WBC and RDW than those without enzyme deficiency. Although each of MCH, MCHC and MCV was higher in the sickle heterozygotes with the enzyme deficiency, when compared with those with the enzyme deficiency, the difference was not statistically significant (P > 0.05). DISCUSSIONThe signs and symptoms of malaria infection in humans are caused by the asexual blood stage of the parasite2. Severe malaria was observed to be complex and could not be accurately represented as a single schema. However, several pathogenic processes could be combined to lead to severe disease such as rapid expansion of infected RBC mass, destruction of both infected and uninfected RBCs and microvascular obstruction. The spectrum of severity ranged from asymptomatic infection in those with acquired and innate immunities to rapidly progressive, fatal illness. In this study, prodromal symptom such as severe headache dominated the malaria presentation. Cohort G6PD non-deficient HbAA subjects (group A) were observed to be more often admitted to hospital for the treatment of malaria than G6PD deficient HbAA (group B), HbAS non-deficient (group C) and G6PD deficient HbAS (group D) subjects. The crude incidence of severe malarial anaemia (Hb < 7 g/dl) and high parasitaemia (> 10,000 parasites/ml) episodes, cerebral malaria episode, hypoglycaemia, impaired consciousness, seizure, respiratory distress, severe headache, fever, diarrhoea and vomiting were higher in group A compared to groups B, C and D subjects. The fact that none of the HbAS subjects suffered cerebral malaria could suggest that HbAS protects against cerebral malaria and coma. Since mortality caused by P. falciparum infection was related to haemoglobin concentration and parasite density21, more HbAS and G6PD deficient HbAA subjects had lower mortality rate from the malaria compared to G6PD non-deficient HbAA individuals. The severe anaemia episodes were more predominant in children below 5 years old while the cerebral malaria episodes although few cases were predominant in older children supporting the observation of Snow et al.22. The severe anaemia observed in this study could neither be drug induced since quinine sulfate (200mg) the mainly used antimalarial drug has not been implicated in haemolysis in G6PD deficiency, nor food-induced, since fava beans is not consumed in the study area. Severe oxidative haemolysis and exercise related rhabdomyolysis from co-inheritance of G6PD deficiency and HbAS were also not observed in this study area. Anaemia became worse after treatment began in some group A subjects, probably due to the high parasitaemia. The anaemia was typically normocytic and normochromic in some though, some expressed a microcytic and hypochromic picture implicating other causes for the anaemia such as iron deficiency. Respiratory distress, defined by deep and grasping breathing was common in groups A and B subjects compared to groups C and D subjects. The subjects of groups A and B were severely anaemic implicating therefore the massive breakdown of red blood cells during the respiratory distress due to poor oxygen transport. The subjects as well suffered simple febrile seizures and recovered after appropriate treatment. Hypoglycaemic episode, most common in children with severe malaria23 was found to be more common in G6PD non-deficient HbAA subjects compared to G6PD deficient HbAA subjects, G6PD non-deficient HbAS and G6PD deficient HbAS. Hypoglycaemia was evident at malaria presentation and could be due to impaired hepatic gluconeogenesis and increased consumption of glucose in peripheral tissue as well as by parasite. Gluconeogenesis probably failed to compensate, in the presence of decreased glycogen flux of glucose, increasing the risk of hypoglycaemia in falciparum infected subjects as reported by Dekker et al.24. The significantly lower (P < 0.05) Hb concentration, PCV, RBC, MCHC and in G6PD non-deficient HbAA subjects during the progress of malaria when compared with those of G6PD deficient HbAA, G6PD non-deficient HbAS and G6PD deficient HbAS is indicative of less severe malarial anaemia in the later groups. The low platelet count in G6PD non-deficient HbAA subjects compared to that in G6PD deficient heterozygotes is consistent with the finding that platelets could form ‘clumps’ with Plasmodium-infected erythrocytes, hence thrombocytopenia may be helpful as a sensitive but not specific marker of active infection25,26 . However, low amount of platelets may not only be a marker of parasite burden but may be protective against severe disease. The higher WBC, lymphocyte and granulocyte counts in group A subjects compared with those of the other groups during the progress of malaria could be associated with severe or acute malaria, consistent with the report of Molyneux et al.23. Group A subjects suffered malaria more frequently, had significantly higher parasite density and percentage parasitaemia (P < 0.05) than groups C and D subjects but not group B. The anaemia herein couldn’t have been due to the G6PD deficiency status since subjects avoided consumption of oxidant drugs with high redox potential. The intraerythrocytic malaria parasite is hypothesised to digest haemoglobin to supply the amino acids needed for the synthesis of their own proteins27. The high parasitaemia observed in groups A and B subjects therefore, could reflect the high consumption of host haemoglobin hence anaemia. Excess haemoglobin consumption by Plasmodium falciparum is said to represent an essential evolutionary strategy to prevent the premature haemolysis of the highly permeabilised infected red blood cells28. Red blood cells with low G6PD activity offered a hostile environment to parasite growth and thus advantage to G6PD deficient subjects resulting in the low parasitaemia observed in group D and B subjects. It is observed that sickle heterozygotes and G6PD deficient individuals experienced reduced fatality and severity of malaria disease. Severe vomiting, diarrhoea and impaired consciousness were all characteristics of algid malaria involving other organs of the viscera. However, it never was as lethal as cerebral malaria in which obstruction could block glucose flow to the brain and since the brain cell depends solely on glucose, death could occur. Black water fever in which there is sudden appearance of haemoglobin in urine indicating severe intravascular haemolysis leading to haemoglobinaemia and haemoglobinuria was common to groups A, B and C but not to D. However, Plasmodium falciparum might not be directly involved in the acute lysis as irregular use of quinine as chemoprophylaxis could also be associated with black water fever. The rupturing of about 5-10% of parasitised RBCs in malaria might not be sufficient to explain the severity of anaemia observed. Severe anaemia may also arise from multiple poorly understood processes including acute haemolysis of infected and uninfected RBC (probably induced by autoimmune response), together with loss of iron (Fe) through hemozoin and dyserythropoiesis, as well as through the interaction of malaria infection with other parasitic infections and nutritional deficiencies14. The clinical features of malaria were diverse in severity and syndrome. However, severe anaemia, hypoglycaemia and impaired consciousness were more common to group A subjects than other groups, though the mechanism remains elusive. Since G6PD is a membrane bound enzyme, HbS/membrane interaction might act as primum movens for the diverse cellular mechanisms which reduce falciparum invasion, impair the parasite survival and development within the red blood cells and accelerate infected erythrocyte clearance by phagocytosis. Consequently, the severity and clinical symptoms of falciparum malaria are ameliorated. However, these two genetic defects are likely playing different roles in clinical P. falciparum malaria. Sickle haemoglobin seems to be a more powerful malarial resistant factor than G6PD deficiency. It could therefore be concluded that inheritance of sickle and G6PD deficiency genes is beneficial since inheriting them protects against severe malaria. G6PD deficiency and HbS in HbAS may be acting as natural vaccines against severe Plasmodium falciparum malaria. Acknowledgement: We thank the North West Provincial Delegate of Health for his permission to carry out this research in his area of jurisdiction. We are obliged to all who volunteered to take part in the study. REFERENCES
© 2006 Nigerian Society for Experimental Biology. The following images related to this document are available:Photo images[bk06014t2.jpg] [bk06014t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}