 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
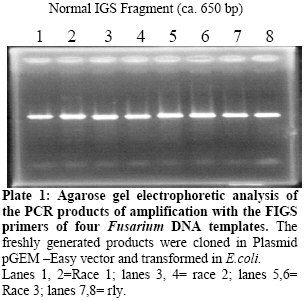
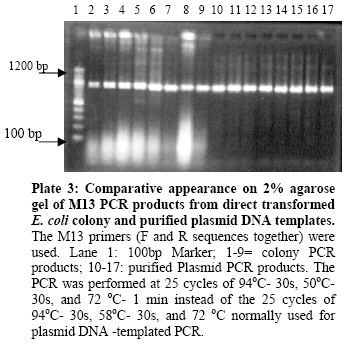
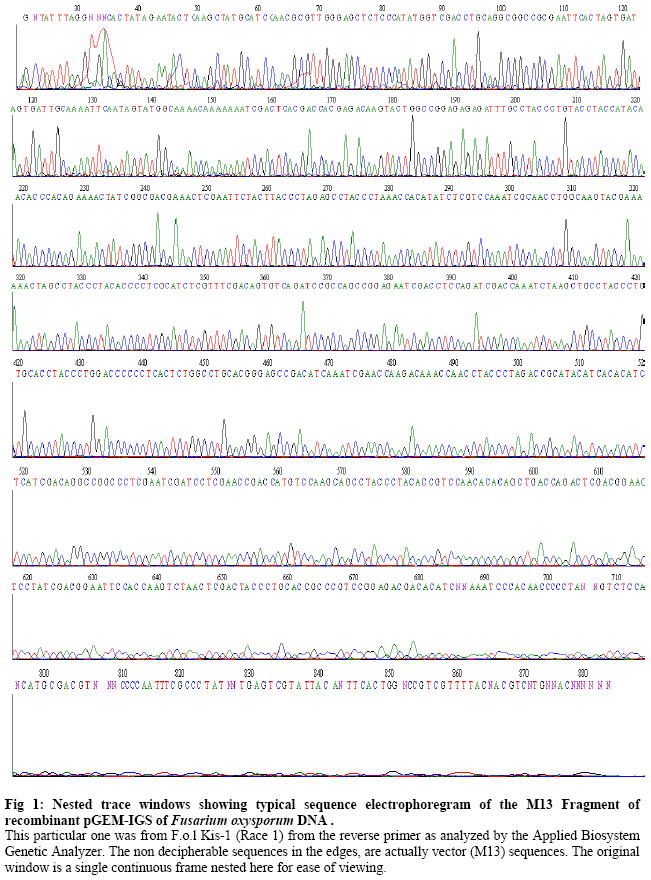
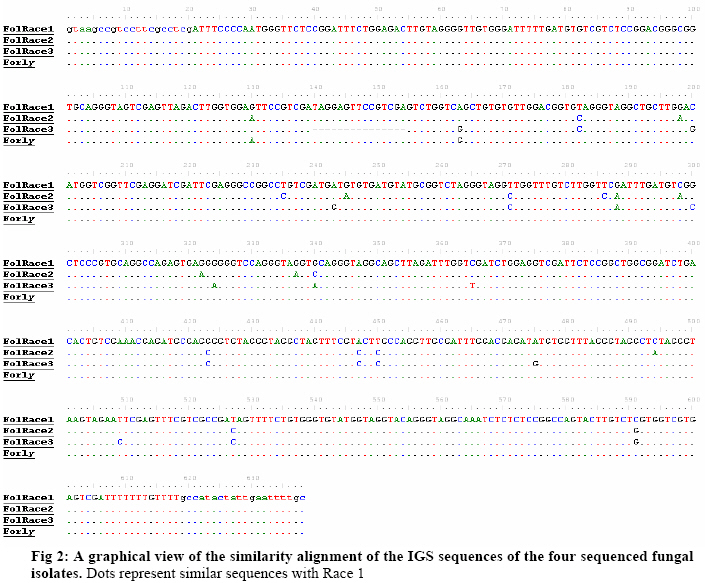
Biokemistri, Vol. 19, No. 1, June, 2007, pp. 1-8 Comparison of Fusarium oxysporum fsp lycopersici races 1, 2 and 3, and f.sp radicis lycopersici based on the sequences of fragments of the ribosomal DNA intergenic spacer region Olusegun Samuel Balogun Laboratory of Plant Pathology, Department of Crop Protection, Faculty of Agriculture, University of Ilorin, PMB 1515 Ilorin, Nigeria, E-mail: samcleo1@yahoo.com Tel: +234-8035814131 Received 17 March 2007 Code Number: bk07001 Abstract Sequence analysis of genomic fragments from the intergenic spacer region from three isolates of Fusarium oxysporum fsp lycoperisci and fsp radicis lycopersici was carried out using the big dye terminator sequencing procedure. Two conditions of the DNA templates were also evaluated for their influence on the outcome of the terminator reaction. Results showed that sequencing using the PCR products of M13 primer reaction with either direct E. coli colony, (condition 1) or purified plasmid DNA as templates (condition 2), were successful and the sequences of the cloned IGS fragments were the same indicating that time and cost could be minimized by excluding the plasmid purification steps. Based on the sequence analysis of the IGS fragment of race 1 (kis-1a) (ca. 638 bp including the forward and reverse primers sequences) it is observed that there is at least 95% similarity between the F. oxysporum races 1, 2, 3, and rly. Using the BioEdit sequence analysis program, there are 14 conserved regions with the longest continuous consensus segment being between nucleotide position number 1 and 129. Region 2 has 18 segment length (164-181), while region 3 is the shortest region with 15 segment length (183-197). Keywords: Fusarium oxysporum, big dye terminator, plasmid DNA-PCR, colony–PCR, sequencing reaction INTRODUCTION The species of Fusarium have traditionally been differentiated by their morphological characteristics on selective media1,2. It is almost impossible, however, to identify pathogenic types, or forma speciales and races of Fusarium oxysporum using morphological features. An inoculation assay using tester plants has been a popular approach of identification of forma speciales. However, this is a time-consuming approach3; thus necessitating development of other methods. Arie et al.4,5 have proposed immunoassys as alternative methods while recently, molecular markers have become popular for identifying species and subspecies in fungi. Some of the techniques that have been reported include amplified fragment length polymorphisms (AFLP)6, random amplified polymorphic DNA (RAPD)7, restriction fragment length polymorphisms (RFLP)8; direct amplification of length polymorphism9 among others. The sequence of DNA encodes the necessary information for living things to survive and reproduce and different organisms are known to have different arrangement of the nucleotides of their DNA. As noted by a contributor in the free on-line encyclopedia -Wikipedia10because of the key nature of DNA to living things, knowledge of DNA sequence should come in useful in practically any biological research. Differentiation of the Fusarium species/subspecies based on comparison of DNA sequences of the ribosomal DNA (rDNA) and internal transcribed spacer (ITS) regions have been reported11. More recently, Hirano and Arie12 have reported differentiation of Fusarium oxysporum f.sp lycopersici and f .sp radicis lycopersici by a polymerase chain reaction (PCR)-based method using specific primer sets developed from the knowledge of the partial nucleotide sequences of the endo (pg1) and exo (pgx4) polygalacturonases genes of the fungi. Based on the sequences of the rDNA intergenic spacer region, endo polygalacturonase gene (pg1) and the mating type genes (MAT1-1-1 and MAT1-2-1), Kawabe et al13 constructed phylogenetic trees for Fusarium oxysporum f.sp lycospersici isolates. They found out that although there was no correlation between races and phylogeny based on rDNA-IGS, pg1 and mating type genes world wide, there was correlation among Japanese isolates. In this study, sequencing the aforementioned 4 fungal isolates is part of basic research to compare and contrast them. It was also aimed at evaluating the influence of condition of DNA template on the sequencing terminator reaction and eventual sequence analysis. MATERIALS AND METHODS Evaluation of the effect of different conditions of DNA templates on big dye terminator sequencing reaction Sequencing reactions were carried out using the big dye terminator sequencing procedure, which is an alternative to the Sanger chain terminator sequencing. The principle behind this procedure is that each of the dideoxynucleotide chain-terminators is labeled with a separate fluorescent dye, which fluoresces at a different wave length10. Recombinant plasmid DNAs were initially generated by separately cloning freshly derived IGS- PCR fragments of the 4 fungal isolates into pGEM –Easy vector and transforming them in competent E. coli cells using the standard procedure14. The IGS fragments that were later sequenced were then derived either directly from the unpurified transformed E. coli colony (plasmid + IgS fragment +E. coli cells) or from the purified recombined plasmid DNAs (Plasmid DNA+ IGS fragment). In both cases, the final sequencing reaction templates were generated from PCR procedures in which the M13 forward and reverse primers were used together and the two aforementioned sources of IGS fragments were the DNA templates respectively. In the E. coli colony PCR, the template was applied by using sterile toothpick to directly pick just a little portion from the desired colony and then shaking it briefly inside 10µl of the PCR reaction mixture that had already been pipetted into the PCR tubes accordingly. The annealing condition for the colony PCR differed a little bit from the normal condition that applied when purified recombinant plasmid DNA was used as template. The E. coli colony PCR, was performed at 25 cycles of 94oC- 30s, 50oC- 30s, and 72 oC- 1 min instead of the 25 cycles of 94oC- 30s, 58oC- 30s, and 72 oC used for the other PCR. Treatment of PCR products with Exo-SAP nuclease The PCR products obtained from both direct bacterial colony PCR and plasmid DNA PCR were subjected to treatment with Exo-SAP (Exonuclease-Shrimp Alkaline Phosphatase conjugate). To 5 μl of PCR product, 2μl of ExoSap was added and the mixture incubated in a thermal cycler at 37oC for 15 min and 80 oC for 15 min. This was to help remove as much impurity as possible from the products before they were eventually used as templates for the sequencing reactions proper. Other Sequencing PCR conditions Under the first and second conditions, each 20 µl mixture contained 2 μl of Exo-sap- treated M13 PCR products as templates, 3.0 µl of Big Dye buffer; 1.0 µl of either of 0.8 pmol M13 primer forward or reverse sequence; 1.0 µl of Big dye terminator and 13.0μl of MilliQ H2O. The PCR temperature cycling condition in all cases was 1 cycle of 96 oC for 1 min, 25 cycles of 96 oC- 10s; 50 oC -5 s; 60 oC- 4 min, and holding at 4 oC. The reaction was carried out with the Gene Amp thermal cycler. A small portion each of the PCR products was analyzed on 2% agarose gel to confirm success or failure of sequencing reaction while the rest were subjected to purification prior being run in the genetic analyzer machine. Purification of sequencing PCR products Ten (10) μl PCR products obtained as described above were transferred to 1.5 ml Eppendorf tubes. To 10μl of the product, 1μl of 3M sodium acetate and 30μl 99.5% Ethanol were added and gently mixed. The mixture was left standing at room temperature for 15 min before centrifugation at 15000 rpm for 20 min. The supernatant was decanted and to the pellet, which contained the desired product, 150μl of 70 % ethanol was added. The mixture was centrifuged for 20 min at 15000 rpm. The supernatant was decanted carefully while the DNA pellet (which is invisible) was vacumn-dried for 10 min using the EYELA evaporator. Ten μl of Hi-Di formamide was added and the mixture vigorously vortexed using the tuple mixer (Iwaki Glass Co. Ltd Japan) to re-suspend the DNA fragments. The products were thereafter kept at 20 oC overnight. Sequencing run and analysis Just before sequencing analysis using the Applied Biosystem 3130x genetic analyzer system, the purified sequencing reaction product was heated for 5 min in boiling water bath to linearize the DNA fragments and transferred immediately on ice to ensure that they remained linear until analyzed. The reaction mixtures, 10 µl in quantity, were transferred to lanes of the MicroAmp optical wells. At the end of the sequencing run, the results in the form of electrophoregrams and deduced textual sequences, were copied out and transferred to a PC where the Genetyx Mac program or BIOEDIT Sequence Analysis program for Windows were used to analyze the sequences based on homology and complementary searches with the Primers sequences and Alignments. RESULTS AND DISCUSSION Plate 1 shows the agarose gel electrophoretic analysis of the IGS-PCR fragments that were cloned into plasmid pGEM -Easy vector prior to transforming in E.coli cells. It shows that they banded around 650 bp. Plate 2 compares the appearance on the gel of the colony PCR products and plasmid DNA PCR products when they were subjected to the same cycling conditions. The important point here is that both contained the target M13 fragments, which encompasses the desired cloned IGS fragment, hence the banding at a position around 900bp. The additional information obtained here was that plasmid DNA template could be amplified at annealing temperature as low as 50oC. Ordinarily, the annealing temperature is 58oC. Plates 3 and 4 show that the desired fragments were amplified in both the direct transformed bacterial colony DNA and purified plasmid DNA-templated reactions. The bands in Plate 3 are produced with M13 forward and reverse primers used together in the PCR reaction, while those in Plate 4 are produced by either of M13 forward or reverse primers respectively. This confirmation was necessary to ensure that the PCR products to be purified actually contained the desired amplicons. Electrophoregram results showed that sequencing run using the PCR products of M13 primer PCR with either direct E. coli colony (condition 1) or purified plasmid DNA as templates (condition 2), were successful and the sequences of the cloned IGS fragments encompassed in them were the same (Fig 1). The implication of this is that the use of direct colony -PCR product as template can cut down on both the time and expenses required to purify the plasmid DNA beforehand when direct colony PCR is employed to generate the PCR product needed for the sequencing reaction. Tables 1, 2, 3, 4 show alphabetical translations (Fasta file format) of the fungal isolates. Races 1, 2, and radicis lycopersici have 638 base pairs each while race 3 had 623 bp. Optimal sequence alignment analysis, and consensus or conserved region search was carried out using the BIOEDIT sequence analysis program for the four isolates. Based on the sequence analysis of IGS of the F.ol race 1, it is observed that there is at least 95% similarity between the F. oxysporum races 1, 2, 3, and rly. The graphical alignment representation is shown in Fig 2. As shown in Table 5, there are 14 conserved regions with the longest continuous consensus segment being between nucleotide position number 1 and 129. Region 2 has 18 segment length (164-181), while region 3 is the shortest region with 15 segment length (183-197). Based on all the facts presented, the ribosomal IGS region was considered representative enough to be used for diagnostic purposes especially in the development of DNA probes that were successfully used in Southern blot analysis to detect the four Fusarium oxysporum DNAs sample preparations (Data not shown here). In fact, based on the sequences of this same IGS, MAT1 and pg1 regions, Kawabe et al.13 had constructed an evolutionary lineage tree of this same tomato wilt pathogen showing that there are 3 three evolutionary lineages, which are each composed of a single mating type and a single or closely related vegetative compatibility group. Acknowledgements The author wishes to express gratitude to the students and staff of the Laboratory of Plant Pathology, Tokyo University of Agriculture and Technology, Fuchu-shi, Tokyo, Japan under the able leadership of Prof. T. Teraoka and Dr. T. Arie for their technical support, and useful pieces of advice. The financial support of the Japan Students Services Organization (JASSO) during the study period is equally appreciated. REFERENCES
© 2007 Nigerian Society for Experimental Biology. All rights reserved. The following images related to this document are available:Photo images[bk07001t4.jpg] [bk07001p3.jpg] [bk07001f1.jpg] [bk07001p4.jpg] [bk07001t3.jpg] [bk07001t2.jpg] [bk07001t1.jpg] [bk07001f2.jpg] [bk07001p1.jpg] [bk07001p2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}