 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Biokemistri, Vol. 23, No. 3, Dec, 2011, pp. 98-107 Review Article Gaining notoriety through translocation: a case of the apoptosis-inducing factor (AIF) in poly (ADP-ribose) polymerase (PARP)-1-dependent neuronal death Amos A. Fatokun* Institute
of Cell Signalling, School of Biomedical Sciences, University of Nottingham
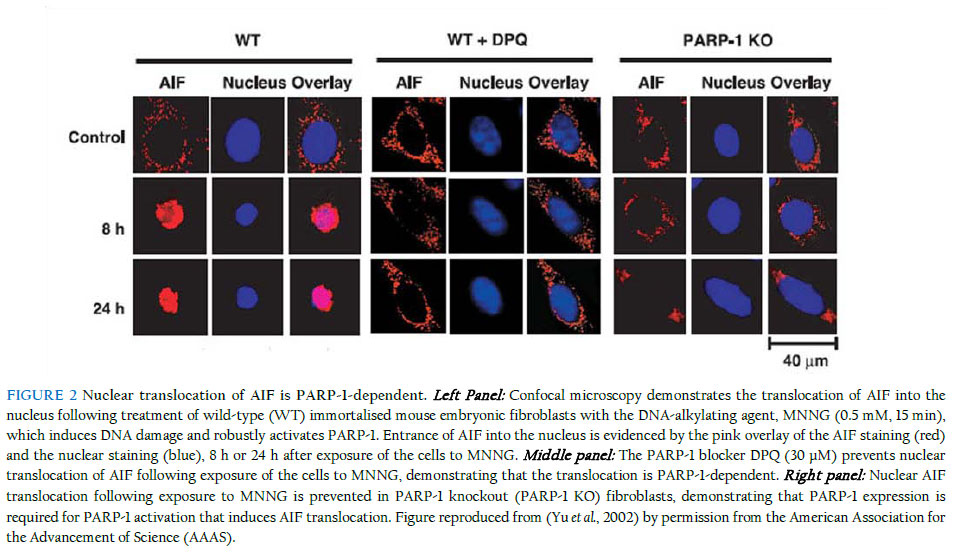
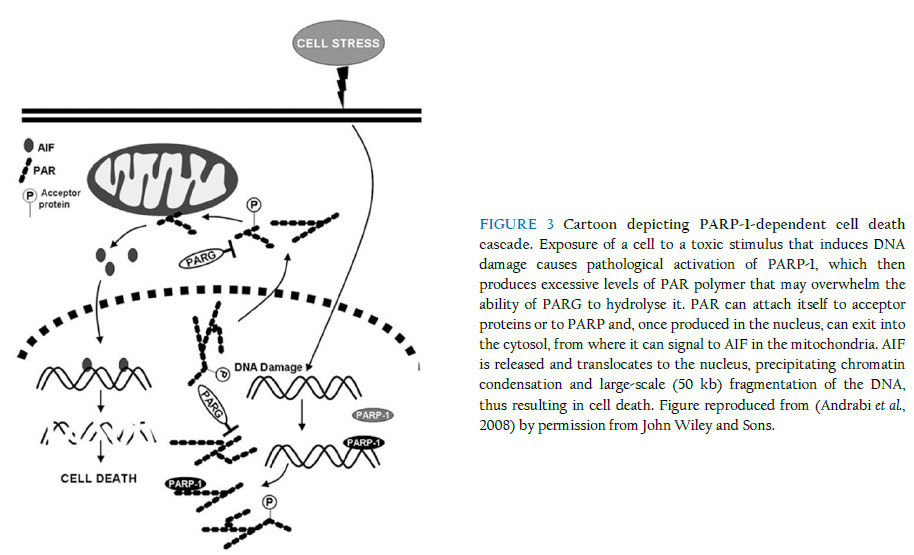
Medical School, Queen’s Medical Centre, Nottingham NG7 2UH, UK Received: 31 December 2011 Code Number: bk11014 ABSTRACT The enormous global burden of neurological and neurodegenerative diseases compels a frantic search for novel neurotherapeutics that will address the underlying pathologies, unlike most of the current pharmacological interventions that predominantly merely relieve symptoms of disease. A major challenge, however, is that the central nervous system (CNS), in contrast to most other well-studied systems, exhibits significant complexity that perhaps makes it one of the, if not the, least tractable to date, at least on the drug discovery landscape. Nevertheless, in the last few years, giant strides in CNS research have yielded significantly-improved understanding of the molecular underpinnings of the various pathological conditions of the brain, not least those associated with the death of neurones, including Parkinson’s and Alzheimer’s diseases. Accordingly, one pathway now known to be majorly involved in the induction of neuronal death is associated with the nuclear enzyme, poly (ADP-ribose) polymerase-1 (PARP-1), whose excessive activation generates a large amount of its polymer, poly (ADP-ribose) (PAR), which, in turn, causes an otherwise beneficial protein, normally resident in the mitochondria, the apoptosis-inducing factor (AIF), to exit its natural domain and enter the nucleus, where it causes large-scale DNA fragmentation and chromatin condensation, ultimately resulting in neuronal death. This review briefly explores the pathological cascade and suggests why targeting it in drug discovery - especially at the level of nuclear AIF translocation - is a rational and promising approach that may eventually deliver novel drugs for clinical use. In addition, the work illustrates how a clear knowledge of the spatio-temporal dynamics of molecular events is highly critical and, in fact, indispensable to a successful drug discovery and development campaign from the bench to the bedside, while at the same time highlighting the multi-disciplinary nature of the drug discovery enterprise. KEYWORDS: Neuronal death; Poly (ADP-ribose) polymerase-1; Poly (ADP-ribose); Apoptosis-inducing factor; Nuclear translocation The phenomenon of cell death In a sense, part of the homeostatic roles of the body is to carefully maintain a useful balance between the number of cells that are produced and the number of those that are destroyed, consistent with the need for a healthy performance of its expected physiological functions. It is known that in the early stages of embryonic development, especially of the mammalian organism, a prodigious number of cells, well in excess of what may be eventually required, is produced, followed by the necessary physiological sculpting that eliminates the surplus, through genetically-programmed cell death, once the various systems, including the nervous system, have been established (Cory et al., 2003). In the mature organism, a disruption of the somewhat-delicate balance causing a net availability of more cells or less cells than needed for proper functioning at any particular time could result in pathological scenarios leading to the establishment of disease. For example, in cancer, there is an abnormally-excessive proliferation of cells due to the loss of normal cell-cycle control on the aberrant cells. On the other hand, several acute neurological and chronic neurodegenerative conditions of the CNS are characterised by abnormal and significant death of functional neurones, and because these lost neurones are not being replaced, the functions they serve in the brain (or in the spinal cord) are also lost. The CNS as we know it is very complex, and it has been the preoccupation of scientific research to understand why, how and even where cells die in pathological conditions such as stroke, Parkinson’s disease and Alzheimer’s disease. It should be promptly said that although the remit of this review is to examine a particular cell death paradigm in the context of the CNS, the phenomenon of cell death, or even the cell death modality being considered here, is not unique or restricted to the nervous system. Devotion to cell death research is rather, relatively, a mature research focus which has seen giant leaps, especially with the advent of excellent experimental tools to examine the molecular mechanisms that underlie the predisposition of a neurone to injury, dysfunction and ultimate demise. In line with the frantic investigations, there has been a flurry of names, perhaps of nicknames too, describing the different types of cell death that have been observed, based on morphological, biochemical, enzymological and immunological features, considered in isolation or in combination. So several and diverse were the names being churned out with notable speed that the Nomenclature Committee on Cell Death recently attempted to rein the frenzy by establishing recommendations for describing cell death modalities, and it now appears that the use of more precise biochemical characteristics (molecular definitions) to describe cell death subroutines has the pride of place (Galluzzi et al., 2012; Kroemer et al., 2005; Kroemer et al., 2009). Perhaps the study of cell death is one current area of scientific investigation that has witnessed an exceptional level of semantic confusion as regards the choice of an apt name to distinguish any observed death paradigm from the other possibilities. This is not surprising, however, as it is often the case that any scenario of cell death could manifest attributes of the different known paradigms to different degrees, with the most prominent feature(s) determining the eventual nomenclature. Just to mention but a few of the cell death modalities, apoptosis, as dubbed by Kerr nearly 40 years ago (Kerr et al., 1972), represents an active form of cell death that is highly regulated, hence the other name “programmed cell death” that is commonly reckoned as synonymous with it, albeit incorrectly, strictly speaking, as other forms of cell death are also now known to be somewhat regulated, including necrosis, a passive form of cell death that was initially conceived as rather largely random, often accompanied by cell swelling and the disruption of the cell membrane (Lorenzo et al., 2007). One major distinguishing feature of apoptosis as observed over the years is its requirement, in most cases, for the recruitment of cysteine proteases, called caspases, for execution of cell death, from initiation to consummation. Further in the cell death arena, autophagy is regarded in some circles as a form of cell death (autophagic cell death) but in some others as an adaptive stress response which enables the cell to delay its “doom” – intracellular materials are degraded and macromolecular entities are recycled (Debnath et al., 2005; Galluzzi et al., 2012; Klionsky et al., 2010). The distinguishing features of apoptosis, necrosis and autophagy were recently described (Wang et al., 2009a). Besides, as the field currently stands, some other forms of modalities are regarded as atypical, and it is certainly a long list – Wallerian degeneration, entosis, anoikis, entosis, necroptosis, pyroptosis, paraptosis, pyronecrosis and cornification, etc (see (Galluzzi et al., 2012; Kroemer et al., 2009)). Notably, the nature of cell death can be dependent on factors such as the type of insult, its degree and duration, and the cellular context in which injury is engendered by the insult. It is beyond the intent of this review to treat these concepts or cell death types in detail, as a number of relevant literature provides in-depth coverage. At this point, however, it is worthy of mention that a potential avenue of investigation that may attain prominence in the future is the elucidation of critical cross-talks that may exist among the cell death types and how such inter-relationships, if present and of some significant functional consequence, can be usefully exploited for novel therapeutics development. To illustrate this potential using the example of a likely interplay between apoptosis and necrosis, it has been suggested that when energy levels within injured cells are too low, they cannot proceed to death through apoptosis, which requires energy for execution, and therefore may seek an alternative but feasible route for their demise, such as necrosis, which could occur despite low cellular energy levels. Poly (ADP-ribose) polymerase (PARP)-dependent cell death – yet another type? The story of PARP illustrates beautifully the consequences of the biphasic capabilities of an important cellular enzyme, and how what is ideally a beneficial biological entity can unfortunately invoke harm in its attempt to remedy a situation that threatens to go out of hand. PARP is a protein normally resident in the nucleus, with recognised physiological roles in the maintenance of cellular homeostasis and genomic stability, among other functions (Smith, 2001). Actually, PARP is a collective name for a group of about 17 nuclear proteins that share significant homology and enzymatic roles, but to date, one of them, PARP-1 (EC 2.4.2.30) (116-kDa) is the most relevant (Hong et al., 2004), for a reason that will soon be discussed. Other appellations occasionally used for the PARP enzyme include poly (ADP-ribose) synthetase or poly (ADP-ribose) transferase. When a DNA base excision occurs, PARP acts as the sensor for the DNA strand nicks and breaks and thus facilitates repair. The activated PARP, whose activity increases about 500-fold, does this by synthesising its polymer, poly (ADP-ribose) (PAR), which has the capacity to attach itself, through a post-translational process called ribosylation, to several nuclear proteins (histones, topoisomerases I and II, DNA polymerases, DNA ligase-2, high-mobility-group proteins, transcription factors, etc) and even to PARP (Shall et al., 2000; Smulson et al., 2000). Because PARP-1 synthesises more than 95% of the entire PAR polymer, it is adjudged to be the most important of the PARPs, and consequently is the most-widely studied (Dawson et al., 2004). Thus, henceforth in this review we shall refer to PARP-1, although the potential significance of contributions from other PARP isoforms is becoming increasingly explored. PARP-1 is highly conserved among eukaryotes (Virag et al., 2002) and features three major domains, an N-terminal domain with two zinc finger motifs plus a nuclear localisation sequence responsible for DNA-binding, a central auto-modification domain, and a C-terminal catalytic domain housing the nicotinamide adenine dinucleotide (NAD)-binding site and the PAR-synthesising domain (Hong et al., 2004; Kameshita et al., 1984). Nature in its smartness seems to have provided a regulatory enzyme, poly (ADP-ribose) glycohydrolase (PARG), which controls PAR levels, following synthesis by PARP-1, through catalytic (hydrolytic) degradation to free adenosine diphosphate (ADP)-ribose units (Davidovic et al., 2001; Whitacre et al., 1995). PARG is encoded by a single gene and localises to the nucleus, but has alternatively-spliced isoforms with diverse patterns of localisation (Bonicalzi et al., 2003; Haince et al., 2006; Meyer-Ficca et al., 2004; Meyer et al., 2007). PARG’s PAR-clearing function has been shown to be directly relevant to PARP-1-mediated cell death, having been found to protect against cell death mediated by the PAR polymer (Andrabi et al., 2006) or other stimuli inducing PARP-1-dependent cell death (Blenn et al., 2006; Cozzi et al., 2006), an observation that further lends credence to the idea that regulating the levels of PAR is highly critical to ensuring protection against the cell death. In a situation where DNA damage is mild, PARP-1 activation leads to repair, but when damage is profound, PARP-1 becomes so over-activated that it produces an exceptionally large amount of the PAR polymer that then begins to contribute to cellular damage and death (Figure 1 shows the time-dependent accumulation of PAR polymer following neuronal challenge with NMDA, which induces robust activation of PARP-1. No detectable levels of the polymer were observed in neurones lacking PARP-1). Interestingly, the cell death elicited by PARP-1 overactivation is unique, although some years back it was mistaken for necrosis (Ha et al., 1999), perhaps due to some features it exhibits that are similar to those of necrosis. In contrast to apoptosis, PARP-1-mediated cell death is largely independent of the recruitment of caspases (Yu et al., 2002), does not involve formation of apoptotic bodies, and precipitates large-, rather than small-scale, DNA fragmentation (Wang et al., 2009a). Although it also involves the loss of membrane integrity, it does not bring about cell swelling, unlike necrosis (Wang et al., 2004; Yu et al., 2002). PARP-1-dependent cell death: biochemical nature, context and players As mentioned earlier, the process that leads to PARP-1-dependent cell death begins by the excessive activation of the nuclear enzyme, induced by exposure of cells to a severe insult that causes appreciable DNA damage. The sequence of biochemical events then proceeds through the accumulation of excessive levels of the PAR polymer that could induce membrane potential dissipation, mitochondrial depolarisation and further downstream pathological events culminating in death, as discussed later. This process occurs in most mitotic and post-mitotic cells in mammalian organisms, occasioned by many different (sources of) toxic insults, including glutamate (especially NMDA) receptor overactivation (in neurones), oxidative stress, nitrosative stress, ischaemia, hypoxia, hypoglycaemia, inflammation and DNA alkylation (Fatokun et al., 2008; Pacher et al., 2008; Stone et al., 2002; Zhang et al., 1994), and relevant to a myriad of diverse pathological situations, including neurological conditions (e.g., stroke and traumatic brain injury), neurodegenerative diseases (e.g., Parkinson’s disease, Alzheimer’s disease, Huntington’s disease and amyotrophic lateral sclerosis (Lou Gehrig’s disease, especially in American parlance)), diabetes, arthritis and liver toxicity (see (Dawson et al., 2004; Virag et al., 2002)). So central now in our current understanding of this cell death paradigm is the role of PAR that a world-class group of investigators has named it “parthanatos” (a bonus to Greek mythology enthusiast?). It is a marriage of two words, “par,” for the poly (ADP-ribose) polymer, and “thanatos,” the personification of death in Greek mythology (Andrabi et al., 2008; Harraz et al., 2008). Various lines of argument have been adduced in an attempt to explain the exact mechanisms involved when cells die at the hands of PARP-1 following its overactivation. Historically, the depletion of NAD+ that follows PARP-1 overactivation causes the affected cells to seek replenishment for the all-important co-factor in order to maintain normal cellular metabolic processes. Unfortunately, however, this drive for replenishment is an energetically-expensive process, since ATP is needed to produce NAD+. It is then conceivable that over time the cells become starved of ATP and so will have to suffer an inevitably suicidal fate (hence the name “suicide hypothesis”), but through a cell death process that does not require the maintenance of a significant energy level, which in this case is necrosis and not apoptosis (Berger et al., 1986; Ha et al., 1999). Well, as logical and attractive as this school of thought may be, it remains to be conclusively determined whether energy depletion plays a major causative role in PARP-1-mediated cell death, although arguments abound both against and in favour of it, and some latest renaissance in examining the role of bioenergetics in cell death may demand a re-think of the current convictions (Liu et al., 2009; Sasaki et al., 2009; Yang et al., 2007). In part, the weakness of the suicide hypothesis relates to its apparent lack of explanation for why it takes so long after the NAD+ has been depleted before the cells (neurones in our case) eventually die. Another assault against the hypothesis comes from observations in PARP-1 knockout mice that their energy stores (levels) following focal ischaemic injury are no different from those of their wild-type counterparts, despite the fact that their infarct volumes are smaller (Goto et al., 2002). These and other grounds for disputation have informed the current quest to look further afield for potential mediators that may be “fit for purpose” in delineating the molecular mechanisms underpinning PARP-1-mediated cell death. Apoptosis-inducing factor (AIF) as a credible mediator of PARP-1-dependent cell death There is now a compelling weight of evidence supporting the responsibility of the mitochondrial flavoprotein, AIF (Susin et al., 1999), for mediation of PARP-1-dependent cell death. Again, the name that this protein still bears is a vivid reflection of how progressive scientific understanding could, and should, be, in an attempt to explore a biological target with previously unknown function(s). No one would have called this protein AIF if we had the knowledge about it when it was first christened as we do today, since it’s been clearly shown that the protein does not induce classical apoptosis, unlike previously thought. Anyway, perhaps for the mere purpose of avoiding any confusion likely to arise from a re-christening, this molecule has been fated to bear “guilt by nomenclature” for a crime there is no convincing evidence it ever commits. The AIF gene is on chromosome X (Susin et al., 1999), synthesised in the cytoplasm (67 kDa) before its importation into the mitochondria where it is processed to a mature 62-kDa form, while its truncated version is 57 kDa (Cao et al., 2007; Otera et al., 2005). Featuring a strong and positive electrostatic potential on its surface as revealed by its solved crystal structures (Mate et al., 2002; Ye et al., 2002), AIF has three domains with distinct functional roles: one on the N terminus that binds flavine-adenine dinucleotide (FAD), another in the centre that binds reduced NAD (NADH), which governs its oxidoreductase activity, and a third, on the C terminus, that underpins its ability to participate in cell death (Susin et al., 1999). It exists in multiple isoforms in humans (Delettre et al., 2006a; Delettre et al., 2006b; Lorenzo et al., 2007) and its oxidoreductase activity is not needed for induction of cell death (Ye et al., 2002). As with PARP, AIF performs physiological roles when resident in its native domain, the intermembrane space of the mitochondria (Susin et al., 1999), although there is recent evidence that a small proportion may also be found in the outer mitochondrial membrane (Yu et al., 2009). The use of the harlequin (Hq) mice in which AIF is down-regulated as much as 80% (Klein et al., 2002) has revealed that, when in its “ordained” environment, AIF supports cell survival, among other possible roles, by mopping up reactive oxygen species (ROS), as evidence was found in these mice of significant oxidative stress and the progressive degeneration of neurones terminally differentiated in the cerebellum and retina (Klein et al., 2002). The culpability of AIF stems from its translocation from the mitochondria to the nucleus, which occurs at the instance of the excessive levels of PAR produced following cellular or tissue damage. Although PARP-1 as the factory for PAR production does not exit the nucleus, its product (PAR) can make a nuclear exit, travelling as far as possible (to the cytosol) to signal, in a manner that remains to be fully resolved, to the AIF in the mitochondria and thus more or less “forcing” it to translocate into the nucleus (Andrabi et al., 2008). The appearance of AIF in the nucleus leads to large-scale DNA fragmentation and chromatin condensation, ultimately precipitating death (Yu et al., 2002). It is now evident that the event of nuclear AIF translocation represents a point of no return for an injured neurone, beyond which recovery or rescue is highly unlikely (Dawson et al., 2004; Hong et al., 2004; Wang et al., 2003). Furthermore, PAR polymer is now considered a definitive signalling molecule bridging the initial process of PARP-1 overactivation and the succeeding event of AIF translocation, with studies having demonstrated that direct exposure of neurones to PAR, even exogenously, can induce AIF translocation, and that toxicity resulting from the exposure correlates positively with the dose and complexity of the polymer (Andrabi et al., 2006; Yu et al., 2006). However, how AIF causes neuronal death following a successful nuclear entry remains to be unequivocally resolved. Possible mechanisms include its binding to the DNA, its recruitment of endogenous proteases or nucleases such as cyclophilin A and endonuclease G (EndoG) (as AIF itself has no intrinsic endonuclease activity) (Cande et al., 2004; Irvine et al., 2005; Wang et al., 2002; Zhu et al., 2007a), or its mediation of DNA vulnerability to such molecules through its interactions with the DNA. Beyond apoptosis-inducing factor: partners in conspiracy As might be suspected, there is now growing evidence of the importance of protein-protein interactions in the dissemination of PARP-1-mediated neuronal death. PARP-1 itself is known to regulate a myriad of proteins and transcription factors, including chemokines, cytokines, induced nitric oxide synthase (iNOS), nuclear factor kappa-B (NF-kB) and signal transducer and activator of transcription-1 (STAT-1) (Kraus et al., 2003; Peralta-Leal et al., 2009). Likewise, the PAR polymer interacts with several proteins, especially those associated with cell death, such as p53 and caspases (Pleschke et al., 2000; Vaziri et al., 1997). In fact, AIF itself has been shown to be a PAR-binding protein through SDS-PAGE analysis, following immunoprecipitation with anti-PAR antibodies, of PAR-associated proteins from PARG-silenced SK-N-SH cells treated with MNNG, with proteins identified through liquid-chromatography tandem mass spectrometry (LC-MS/MS) (Gagne et al., 2008). Drawing from the aforementioned, one of the lingering conundrums to date is whether a number of downstream effectors are responsible for linking PAR signalling with AIF release or AIF-mediated neuronal death. In this context, some proteins, mitochondrial or otherwise, have been implicated to interact with AIF at various signalling levels, with potential consequences for PARP-1-dependent neuronal death, and they include the Bcl-2 proteins, calpains, cathepsins, and heat-shock protein 70 (HSP70) (Bano et al., 2005; Cao et al., 2007; Cheung et al., 2005; Moubarak et al., 2007; Polster et al., 2005; Vosler et al., 2009; Wang et al., 2009b)(Chaitanya et al., 2008; Gurbuxani et al., 2003; Ravagnan et al., 2001), but for most of these molecules there is yet paucity of evidence, or there is conflicting evidence so far, to argue their definitive or direct involvement in AIF release, although such potential influence(s), if confirmed, may complicate the untangling of the entire pathological profile of PARP-1-mediated neuronal death. Some models for the study of PARP-1-dependent cell (neuronal) death A number of experimental conditions have been shown to reproduce features of PARP-1-mediated cell death and are therefore currently being employed to study its mechanistic details and also as basis for screening assays for the identification of small molecules that could intercept the pathway. In cell lines such as HeLa and CHO, or in mouse or human embryonic fibroblasts, the DNA-alkylating agent N-methyl-N'-nitro-N-nitrosoguanidine (MNNG) induces PARP-1-dependent cell death in a highly-specific and reproducible fashion, and this effect could be blocked significantly, if not completely, by PARP-1 inhibitors, but not by caspase inhibitors (David et al., 2006; Ethier et al., 2007; Keil et al., 2006; Yu et al., 2002). Figure 2 illustrates the nuclear translocation of AIF in MNNG-treated wild-type fibroblasts and the inability of AIF to translocate in fibroblasts treated with MNNG in the presence of the PARP-1 inhibitor DPQ or in fibroblasts from PARP-1 knockout animals treated with MNNG. For examination of the cell death pathway in a neuronal context, stimuli that can induce toxicity in primary dissociated neuronal cultures, such as N-methyl-D-aspartate (NMDA), an agonist acting at the NMDA receptor (one of the three ionotropic glutamate receptors), or exposure of neurones to ischaemic, hypoxic or hypoglycaemic conditions, are able to induce the activation of PARP-1 through overactivation of the NMDA receptor leading to PARP-1-dependent cell death following a number of downstream steps, including excessive rise in calcium levels within the cell that activates many calcium-dependent processes and enzymes, including neuronal nitric oxide synthase (nNOS) that causes nitric oxide (NO) release, which may be followed by the reaction of NO with superoxide to produce the highly-toxic peroxynitrite that induces significant DNA damage (Fatokun et al., 2008; Pacher et al., 2008; Stone et al., 2002; Zhang et al., 1994). NMDA and PAR polymer were found to be less toxic to neurones from the Hq mice (which have 80% downregulation of AIF) and restoration of AIF expression in these neurones increased their vulnerability to NMDA-induced cell death to levels found in wild-type neurones (Yu et al., 2006). Consistent with these observations, the Hq mice also displayed reduced lesions, compared to wild-type animals, in experimental models of excitotoxicity and stroke (Culmsee et al., 2005; Yuan et al., 2009; Zhu et al., 2007b). Apart from exposures involving receptor activation, challenging neurones directly with reactive species producing oxidative stress (e.g., hydrogen peroxide) or nitrosative stress (e.g., nitric oxide, peroxynitrite) can also induce DNA damage and lead to PARP-1-mediated neuronal death (Fatokun et al., 2007; Fatokun et al., 2008). However, in order to minimise potential contributions from the other forms of cell death, experimental variables such as concentration of the toxic stimulus and duration of exposure to it should be optimised in order to favour the induction of PARP-1-mediated cell death. It should be noted that whole-animal models of neurological conditions and neurodegenerative diseases and even post-mortem investigations have also been adopted to study PARP-1-mediated cell death, with pharmacological or genetic interventions against PARP-1 reported to be protective in studies that examined them (Komjati et al., 2005; Moroni et al., 2009; Oh et al., 2006; Slemmer et al., 2008; Wang et al., 2003; Yu et al., 2010). Recently, a protein, named “Iduna”, shown to be a PAR-dependent E3 ubiquitin ligase (Kang et al., 2011), was described as the first known endogenous inhibitor of PARP-1-mediated cell death (parthanatos) (Andrabi et al., 2011). Mechanistically, the protective effects of “Iduna” are due to its ability to bind PAR and are thus independent and downstream of PARP-1 activity (Andrabi et al., 2011). Opportunities for therapeutic intervention: blockers of nuclear AIF translocation as novel therapeutic agents It is now firmly established that PARP-1-mediated cell death is unique and distinguishable from other forms of cell death, with nuclear AIF translocation representing a key signature step in the progression from injury to death. The pathway, as explained earlier, seems capable of lending itself to potential therapeutic, and perhaps other, interventions at various levels of the cascade (see Figure 3). Accordingly, attempts have been made to identify chemical molecules that can prevent or, at least, attenuate this cell death, most especially at the level of PARP activation (Gero et al., 2008; Virag et al., 2002), and, as a consequence, there are now series of PARP blockers with varying potencies belonging to different chemical classes and/or generations in drug development. Some have been tested in clinical trials (e.g., INO-1001 (Inotek) for myocardial ischaemia) (Lorenzo et al., 2007), but most are still at the experimental stage. Examples of first-generation PARP blockers are nicotinamide, benzamide and 3-aminobenzamide (3-AB). Second- and third-generation blockers with improved properties (e.g., DPQ and PJ34, respectively) belong to a wide array of chemical classes (Eltze et al., 2008; Peralta-Leal et al., 2009) and some entered clinical trials, although in most cases not for the prevention, but for the promotion, of cell death (in cancer) (see (Rouleau et al., 2010)). This is because the inhibition of PARP-1 in cancer cells will prevent the activation of PARP-dependent DNA damage repair mechanisms in these cells and thus facilitate their death. Interestingly, the broad-spectrum tetracycline antibiotic, minocycline, was found to inhibit PARP activity at nanomolar concentrations (Alano et al., 2006). While the development of PARP inhibitors represents a rational drug-design approach, the identification of chemical entities that can prevent the nuclear translocation of AIF may, in a sense, represent a better therapeutic strategy, especially in relation to achieving the therapeutic goal of preventing neuronal death in neurological and neurodegenerative conditions. Although it may be daunting to be able to find molecules that will act with such surgical specificity, the fact that PARP has a physiological role that should be maintained makes it more desirable to block nuclear AIF translocation (and leave PARP alone to do its beneficial work) that occurs further downstream, rather than block PARP activation, as the latter, if prolonged, could be detrimental to the health of the cells (neurones) that are being rescued. It could therefore mean that PARP blockade may be more relevant to chemotherapeutics, where inhibitors can prevent DNA repair in cancerous cells and thus promote their death. Interactions of AIF with other proteins as it travels from the mitochondria to the nucleus can also be targeted, but this will only be possible after its network of interacting proteins has been sufficiently clarified. Drug discovery efforts in this direction will undoubtedly involve experimental, computational and other approaches, with the availability of the crystal structure(s) of AIF primed to significantly aid computational modelling. Collaborations will span various disciplines or technological platforms, including biochemistry, molecular biology, pharmacology, medicinal chemistry, computational biology and drug discovery. In general, there is no denying the fact that latest trends in tackling biological targets for therapeutic benefit have confirmed or are confirming that the walls that have separated the disciplines for so long are fast tumbling down, and wisdom demands that we embrace the “holistic” approach to bench-to-bedside drug discovery that will occupy the central stage in the 21st century and even well beyond! Summary This work examines succinctly a distinct form of cell death, mediated by overactivation of the nuclear enzyme, PARP, which has now been implicated in the pathophysiology of neurological and neurodegenerative diseases as well as other conditions. The review presents an account of the evolution of scientific understanding of the molecular events by which the cell death is characterised, the associated players in the cascade, and, where already clearly-defined, their spatio-temporal interplay. Blockade of nuclear AIF translocation is then presented as a more promising avenue for therapeutic intervention. Through the exploration, attempt is made to illustrate the challenge of finding, as well as sufficiently characterising, a credible and “druggable” therapeutic target, using a collection of basic biology approaches from different disciplines, before a successful drug campaign is launched, with the hope of finding novel lead molecules that will then be optimised to obtain candidate drugs for testing in clinical settings. It is hoped that such drugs will represent a worthy advancement over existing ones in their ability to directly combat the underlying death of neurones that is the real plague in many neurological and neurodegenerative conditions. Acknowledgements The author gratefully acknowledges the award of a Nottingham Advanced Research Fellowship (NARF) by the University of Nottingham (UK) and of a Marie Curie International Incoming Fellowship (IIF) by the European Commission through its European Union (EU) Seventh Framework (FP7) Marie Curie People Work Programme. References
Copyright © 2011 Klobex Academic Publishers The following images related to this document are available:Photo images[bk11014f2.jpg] [bk11014f3.jpg] [bk11014f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}