 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Culture Collections, Vol 6, No. 1, 2009, pp. 3-9 Virulence of uropathogenic Escherichia coli Georgi Slavchev1*, Emiliya Pisareva2 and Nadya Markova1 1Department of Pathogenic
Bacteria, The Stephan Angeloff Institute of Microbiology,
Bulgarian
Academy of Sciences, 1113
Sofia, Bulgaria;
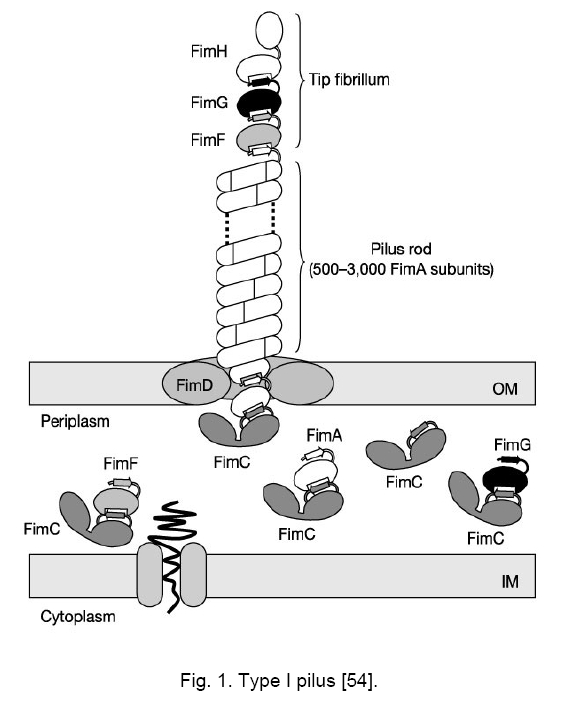
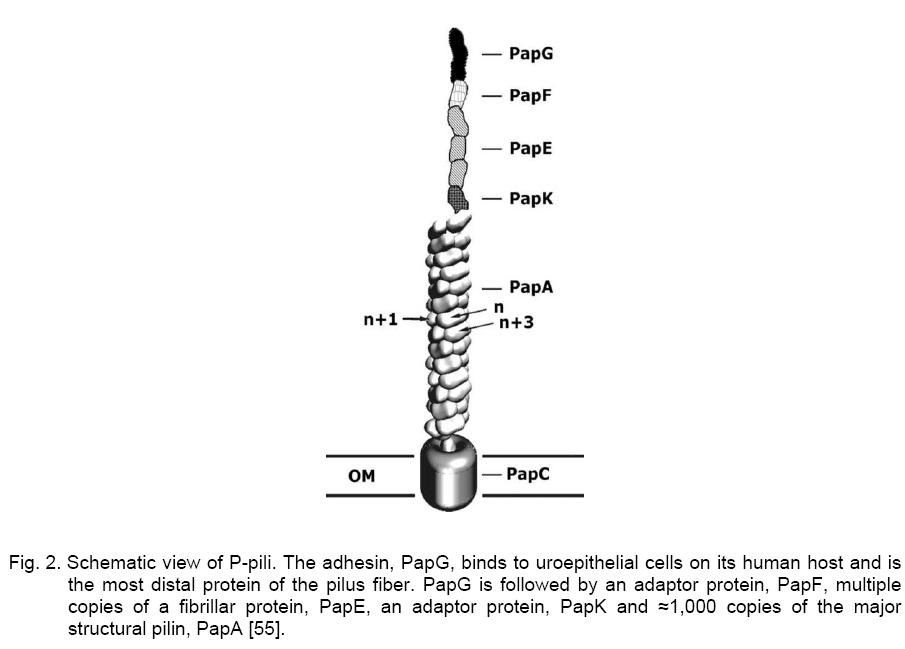
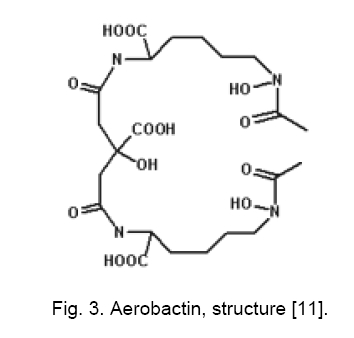
Code Number: cc09001 Summary Uropathogenic Escherichia coli (UPEC) are the most important group of microorganisms responsible for urinary tract infection. UPEC differ from non-pathogenic E. coli and from other E. coli pathotypes by the production of specific virulence factors, which enable the bacteria to adhere to uroepithelial cells and to establish urinary tract infections. Besides adherence factors, toxins, “modulins”, capsules, iron uptake systems and other bacterial products contribute to the virulence of the strains. Urinary tract infection is the most frequently diagnosed kidney and urologic disease and E. coliis by far the mostcommon etiologic agent. Uropathogenic strainshave been shownto contain blocks of DNA termed pathogenicity islands (PAIs) whichcontribute to their virulence. Keywords: Escherichia coli, urinary tract infection, virulence Introduction Escherichia coli, the most prevalent facultative gram negative bacillus in the human fecal flora, usually inhabits the colon as an innocuous commensal. According to the special pathogenicity theory [44], special properties enabling E. coli to overcome host defenses in a new environment, are necessary in order for it to escape the limitations of the colonic mileu and move into new niches devoid of competition from other bacterial species [16, 44]. Uropathogenic E. coli (UPEC) strains are responsible for approximately 80 % of community-acquired and 30 % of nosocomial-acquired urinary tract infections (UTIs). Females under 10 years of age, or between 18 and 40, are at the highest risk for community-acquired infections. Infections in children are often due to blockages in the urinary tract, resulting in pools of stagnant urine. UPEC can reside in the colon and then be introduced into the urethra. Urinary tract infections result from ascending colonization of the urinary tract by these strains. Infections can occur in the urethra (urethritis); bladder (cystitis), moderately severe; associated with burning and pain on voidingplus, possibly, suprapubic pain or tenderness from bladder inflammation and kidneys; (pyelonephritis) - most severe; associated with fever, chills, and flank pain from renal inflammation. In order to colonize and establish a UTI, UPEC strains take advantage of assortment of virulence properties [28, 50]. Bacterial adherence to and colonization of the urinary tract by UPECstrains are mediated by the expression of several types of fimbrial and nonfrimbrial adhesins [21, 23], like most adhesions, it has been difficult to precisely define the role of any particular adhesine due to overlapping function. Type I and P fimbrie, the most common fimbrie found in UPEC strains enhance virulence and are involved in initial urethral colonization. Many UPEC produce hemolysin, which may be involved in kidney disease. Certain UPEC strains posses iron sequestration systems to assist in growth; others produce a capsule that may help avoid clearance from the urinary tract. Virulence factors (VFs) 1. Adhesion Adherence to solid substrates is a property common to many pathogenic microorganisms, including viruses, gram-positive and gram-negative bacteria [5]. By attaching to host structures, microbial pathogens avoid being swept along by the normal flow of body fluids (blood, urine, intestinal contents) and eliminated, although host cells with adherent bacteria can be shed, thereby eliminating the organisms despite attachment [43]. Adherence of E. coli isolates to uroepithelial cells is used to differentiate between uropathogenic and fecal strains [48, 49]. These authors suggest that uropathogenic strains may present a mean of 20 bacteria/cell or more, while fecal strains may present a mean of about 7 bacteria/cell. UPEC strains causing UTI typically agglutinate human erythrocytes despite the presence of mannose (mannose-resistant hemagglutination [MRHA]) [15, 17] and adhere to human uroepithelial cells [47, 51]. Also, adherence to uroepithelial cells is usually unaffected by mannose (mannose-resistant adherence) and is more common among strains exhibiting MRHA than among those exhibiting only mannose-sensitive hemagglutination. Type I pili. Type I pili, originally identified by their ability to mediate mannose-sensitive agglutination, are expressed by the majority of UPEC strains derived from patient with cystitis and pyelonephritis, their role in disease is controversial. Type I pili play a role in gastrointestinal disease, although many enteric E. coli pathogens and nonpathogenic fecal isolates produce this type of pili. Adherence mediated by type I fimbriae is blocked by solutions of D-mannose or α-methylmannoside and by concanavalin A (a lectin that binds to mannoside residues) but not by solutions of other monosaccharides or their derivatives [20, 42]. The type I-fimbrial receptor is probably an extended structure, since mannosides with an α-linked aromatic group, trisaccharides, and branched oligosaccharides that contain (α-1-3) - linked mannosides are better inhibitors of type I-fimbrial binding than D-mannose or α-methylmannoside alone [19, 39]. Receptors for type I fimbriae are present on erythrocytes from many species [15]. Type I-fimbriated bacteria adhere to human buccal epithelial cells [4], intestinal cells [26], and vaginal cells, suggesting a possible role for type I fimbriae in E. coli colonization of the mouth, gut, and vagina. Receptors for type I fimbriae are present in blood vessel walls and in the muscular layers but not the epithelium of the human bladder. Type I pili have composite structure of 7 nm in width and 1 - 2 microns in length and consist of long rigid rod and a distal thin fibrillar structure [29]. E. coli fimbriae, type I fimbriae are encoded by a gene cluster fim that includes genes for a structural subunit, an adhesin, several accessory proteins (involved in subunit transport and assembly and in anchoring assembled fimbriae), and regulatory proteins (Figure 1) [1, 3]. FimH is the adhesin protein responsible for binding to mannosylated glycoproteins and is located at the distal tip of the heteropolymeric type I pilus rod, which is predominantly constituted by FimA subunits [2]. FimG and FimH are minor components that are probably needed as adaptors, initiators or terminators. FimC is the chaperone and FimD outer membrane usher [32]. FimH mediates E. coli binding to mannose-containing glycoprotein receptors - uroplakins, which are located on the luminal surface of the bladder epithelial cells [38]. PAP. P pili (pyelonephritis-associated pili, PAP) are encoded by pap genes found within pathogenic islands. This adhesin is critical for upper UTI infections. Two molecules guide newly synthesized pilus components to the bacterial surface (Figure 2) [25, 53]. PapD transports several pilus subunits from the cytoplasmic membrane to the outer membrane [24]. The complexes PapD - subunit are targeted to the PapC outer membrane usher, which forms a pore through which the pili are translocated across the outer membrane. The major subunit of the pili is PapA anchored in the outer membrane by PapH, PapH terminates fimbrial assembly and helps anchor fimbriae [6]. PapG is the actual molecule which mediates adherence. It is joined to the PapE tip fibrillum by the adapter protein PapF (Figure 2). PapF is also important in the initiation of subunit polymerization. PapG binding to Gal-Gal receptor moiety follow expression of AirS sensor-regulator protein, thereby allowing activation of the UPEC iron- acquisition system. Efficient iron-acqui-sition allows UPEC to grow in urine, an other-wise growth-limiting environment. P-fimbrial expression is subject both to rapid, random phase variation and to environmental influences. Glycolipids containing the Gal-Gal moiety are receptors for adhering E. coli strains and that this moiety is the major determinant of binding. Receptors for P fimbriae are present on erythrocytes from humans, pigs, pigeon, fowl, goats, and dogs but not on those from cows, guinea pigs, or horses [36]. The distribution of these receptors differs among hosts and tissues, and differential expression of the PapG adhesin at the pilus tip could determine tissue and host specificity. Although Gal-Gal-containing glycolipids are only a minor component of the membrane glycolipids of shed human uroepithelial cells, they are the predominant species of human renal glycolipids [36, 37]. The prevalence of P fimbriation in isolates from patients with bacteremia arising from a UTI (urosepsis) is in isolates from pyelonephritis patients. P fimbriae also bind to the epithelial and muscular layers of the bladder [31] and to a loosely adherent surface-associated substance on human colonic cells, possibly contributing to E. coli colonization of the human intestinal tract. P fimbriae are important in the pathogenesis of UTI, primarily because they mediate Gal-Gal-specific bacterial adherence to epithelial cells within the human urinary tract, thereby permitting bacterial colonization and stimulating inflammation. In compromised hosts the requirement for P fimbriae in initiating serious UTI is decreased, suggesting that P fimbriae are necessary for E. coli to overcome certain components of the normal host defense system. Afimbrial adhesins. E. coli produce a family of adhesins that bind to the same mammalian receptor. This family includes F1845 and Dr, which are fimbrial adhesins found in gastrointestinal pathogens, and two nonfimbrial adhesins (Afa-I and Afa-III) expressed by many UPEC [9, 34]. All of these adhesins use as receptor the Dra blood group antigen present on decay accelerating factors (DAF) on erythrocytes and other cell types. Different strains in the Dr adhesin family appear to recognize different portions of the Dr antigen. The adhesin gene clusters of different members of the Dr family are organized similarly, with five closely spaced genes, including one for the structural hemagglutinin [33, 41]. 2. Hemolysin The cytolytic protein toxin secreted by most hemolytic E. coli strains is known as alpha hemolysin [12]. Approximately half of UPEC strains that cause upper UTIs, about a third of those that cause lower UTIs, and only about 10 % of fecal isolates produce a hemolysin (HlyA) that belongs to the repeat toxins (RTX) family. Hemolysin molecules insert into lipid-containing membranes producing cation-selective channels of large conductance with a diameter of 2 nm [8] that increase the permeability of erythrocyte membranes to Ca2+, K+, mannitol, and sucrose [8]. In addition to lysing erythrocytes, hemolysin is toxic to arange of host cells in ways that probably contribute to inflammation, tissue injury, and impaired host defenses. Such toxic activity may contribute to the kidney damage seen in pyelonephritis. Four genes (hlyA, hlyB, hlyC, hlyD)are required for the production and export of the hemolysin. hlyA encodes the hemolysin structural gene. The 110-kDa HlyA protein is unique among E. coli toxins in that it is secreted acrossboth membranes without cellular lysis and without cleavage of asignal peptide [45]. HlyC acylates HlyA posttranslationally, adding fatty acids to two internal lysine residues via amide linkage [46]. The HlyB and HlyD proteins are needed for HlyA secretion out of E. coli, as it the TolC protein. Hemolysin production is associated with human pathogenic strains of E. coli, especially those causing more clinically severe forms of UTI. 3. Aerobactin Aerobactin, a bacterial siderophore, has recently been shown to be associated with E. coli strains which cause pyelonephritis and cystitis [14]. It is an iron sequestration and transport system which enables E. coli to grow in iron-poor environments such as dilute urine andcomplement-depleted serum. In almost all E. coli strains, the aerobactin system is encoded by a five-gene operon, with four genes encoding the enzymes needed for aerobactin synthesis and a fifth gene encoding the outer membrane receptor protein. Aerobactin determinants are found both on plasmids and on the bacterial chromosome, with the chromosomal location predominating among human clinical isolates [27]. Aerobactin production is regulated by the intracellular iron concentration through the fur (ferric uptake regulation) gene product [10, 13]. When iron concentrations are high enough, the fur repressor complexes with iron (or other divalent cations) and binds to an "ironbox" in the promoter region of the aerobactin operon (and other iron-regulated genes), blocking transcription (50). In low-iron conditions, the fur repressor is released from the promoter region and transcription proceeds. Aerobactin is a small molecule (Mw 616) formed from the condensation of two lysine molecules and one citrate (Figure 3) [7]. Following secretion by E. coli cells, aerobactin extracts Fe3+ from host iron-binding proteins and is taken up through a 74-kDa outer membrane receptor protein which is also the receptor for cloacin. The aerobactin system is associated with E. coli isolates from serious UTI and other serious infections in humans and animals, probably because it promotes bacterial growth in the limiting iron concentrations encountered during infection. The chromosomal aerobactin system is associated with other uropathogenic VF determinants, whereas the plasmid aerobactin system is often carried by plasmids encoding multiple antimicrobial agent resistance. The aerobactin receptor protein is a potential target for an antiaerobactin vaccine, and a unique enzymatic step involved in aerobactin biosynthesis could be the target of antiaerobactin chemotherapy [28]. 4. Pathogenic islands
Many of
the virulence genes of pathogenic strains of E. coli are carried in
large multigene chromosomal segments called pathogenicity islands (PAIs) that
are absent from normal fecal and laboratory K-12 strain
of this bacterium. The uropathogenic strain
536 contains two large unstable pathogenicity islands: PAI-I (70 kb) and PAI-II
(190 kb). Both pathogenicity islands are flanked by short (16 - 18 bp) direct
repeats, which are likely responsible for their deletion due to recombination at
a frequency 10-3 [22]. PAI-I encodes the hly hemolysin operon. PAI-II encodes
another hly operon and prf (P-related fimbriae) pilus operon. Uropathogenic
strain J96 contains two pathogenic islands inserted in two different tRNA genes
(pheV at 64 min
for PAI-IV and pheR at 94 min for PAI-V) [52]. The E. coli J96 chromosome encodes several
well-characterized factors involved in extraintestinal disease, including two
types of P fimbriae (Pap and Prs), hemolysin, and the cytotoxic necrotizing
factor 1 (CNF-1) toxin [35]. Previous studies indicated that the P-fimbrial and CNF-1 genes
are linked to the hly operon in strain J96. About one third of UPEC
produce CNF- E. coli CFT073 possesses the smallest of the five PAIs described (50 kb), which is inserted in the vicinity of the metV gene and carries an hly operon [30]. Conclusion Uropathogenic E. coli are the major causative agent of urinary tract infections. UTIs are considered acute, self-limiting infections despite the prevalence of recurrent symptoms two or more times within months of a primary infection. The biological characteristics of uropathogenic E. coli strains include hemolysin and aerobactin production, expression of P fimbriae, serum resistance, cytotoxic necrotizing factor, and capsule production. The currently recognized virulence factors account for only a fraction of the total virulence of wild-type strains, evidence suggesting that other as-yet-unidentified properties that are important determinants of virulence await discovery and characterization. References
Copyright 2009 - National Bank for Industrial Microorganisms and Cell Cultures - Bulgaria The following images related to this document are available:Photo images[cc09001f2.jpg] [cc09001f3.jpg] [cc09001f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}