 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
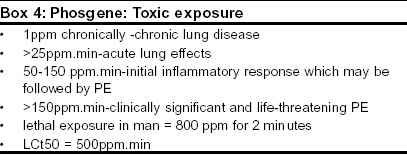
Indian Journal of Critical Care Medicine, Vol. 7, No. 4, , 2003, pp. 250-256 Review Article Pathophysiological aspects of clinical management following toxic trauma Baker DJ SAMU de Paris, Hopital Necker-Enfants Malades, Chairman, ITACCS Toxic Trauma and Hazmat Committee. Code Number: cm03004 Introduction Exposure to toxic agents may occur as a result of accidental or deliberate release. Accidental release of industrial chemicals may produce small scale incidents or mass disasters such as that which occurred in Bhopal in 1984.[1] Deliberate release occurs when toxic agents are deliberately used as weapons of war or by terrorists. The consequences for medical management systems, of both accidental and deliberate release are considerable. This article considers the clinical presentation of a selection of the most common toxic agents from a pathophysiological standpoint. Toxic trauma Systemic attack by toxic agents The respiratory system is the most immediately vulnerable to toxic attack both in terms of structure and function. The end results on the airways are blockage, increased resistance and decreased compliance. Effects on the breathing control system lead to failure of lung ventilation. Actions on the muscles of the thoracic cage have the same effect The production of toxic pulmonary oedema causes a diffusion failure in oxygen passage leading to hypoxaemia. Organophosphate attack on the cholinergic nervous system Atropine, often in high dosage is used to block the muscarinic actions of OP but the alkaloid has no specific action at the voluntary nicotinic synapses. Therapy here depends upon the use of oximes to regenerate the cholinesterase. Compounds used include pralidoxime, obidoxime or the newer Hagedorn oximes such as HI 6.[5] Life -threatening convulsions may be controlled by diazepam. A combination of pralidoxime, atropine and lysine-diazepam is the basis of the military immediate -use injection following nerve agent exposure. Airway clearance and artificial ventilation are a vital part of the management of OP exposure. The development of small gas powered ventilators powered by compressed oxygen has allowed early provision of IPPV of a quality usually associated with hospital care in the pre-hospital area. Ventilatory support is now part of many major toxic response plans.[6] Apart from the acute cholinergic syndrome the OP Intermediate Syndrome (IMS) presents a pathophysiological development following OP exposure which has important consequences for subsequent clinical management. Sennanayake and Karralliede in 1987[7] described a re-paralysis of 10-20% of patients at 18-24 hours post OP pesticide exposure following resolution of the acute cholinergic syndrome There was an apparent change in the nature of NM block with indications of decrementing responses, signifying a non-depolarising block. IMS affects proximal limb, cranial motor and respiratory muscles. The syndrome was confirmed by many authors during the 1990′s with neurophysiological evidence pointing to a post -neuromuscular junctional lesion.[8] Clinical analogies exist for the syndrome including dual block following prolonged succinyl choline, first described when this anaesthetic technique was used during the 1950s and 60s.[9] There are also similarities with myasthenia gravis where there is an immune based down-regulation (reduction in density of AChR at the post-junctional membrane). Clinical neurophysiological studies of IMS[8] have indicated fade at low frequency stimulation and absence of post-tetanic facilitation which point to a post- junctional lesion. More information may be provided from single fibre electromyography (SFEMG), a sensitive neurophysiological technique which records action potentials from two muscle fibres innervated by a common terminal pathway.[10] Analysis of the time variations between the potentials (jitter analysis) gives information about the safety margin of AChR at the NMJ. In conditions where this is reduced such as myasthenia gravis, Eaton Lambert syndrome and administration of non-depolarising muscle blockers such as curare the jitter increases and in some cases leads to a failure of firing of muscle fibres, known as blocking. Significant degrees of blocking are detected as fade following conventional repetitive stimulation but SFEMG will detect changes in neuromuscular transmission long before they are evident using conventional techniques and clinical observation. Baker and Sedgwick[11] reported an SFEMG study of eight fit volunteers exposed to trace amount of sarin (corrected RBC AChE inhibition 40%). The study showed small increased jitter changes seen at three hours post exposure with overall increased jitter changes seen at three days post exposure. No clinical neuromuscular deficit was apparent. Jitter profiles were normal between one and two years post exposure. These results may indicate a sub-clinical change in NM safety margin after non -toxic OP exposure. A number of possibilities have been advanced to explain the origins of the Intermediate Syndrome. These include neuropathy and myopathy together with pathophysiology of the neuromuscular junction itself.[12] Neurophysiological evidence suggests that the NMJ is the critical site and debate centres around whether pre- or post-junctional receptors are involved. At the present time consideration of analogous clinical conditions suggests that it is down regulation of the AChR which is responsible but evidence remains lacking. The third stage of clinical presentation of OP intoxication is organophosphate-induced delayed neuropathy. This is thought to be an action of the OP on the neurotoxic esterase enzyme system and is not related to the anticholinesterase properties of the compound.[13] Botulinum toxin Botulinum toxin acts at the nerve terminal of cholinergic synapses and blocks the release of acetyl choline. After being taken up into the vesicles and translocated to the cytoplasm where the toxin catalyses proteolysis of components involved in the calcium-mediated exocytosis of ACh. The inhibition is permanent and recovery occurs only after the creation of new terminal boutons. The toxin thus blocks neurotransmission, parasympathetic synapses and peripheral ganglia. Conventionally after ingestion of the toxin (the usual route) the parasympathetic action produces a dry mouth , followed by signs of a progressive bulbar palsy (dysarthria, dysphasia and dysphagia) and ocular signs (diplopia and ptosis) Following this there is a progressive symmetrical descending muscular weakness leading to respiratory failure requiring prolonged ventilatory support. NM testing shows a classical presynaptic decremental pattern to repeated stimuli with post-tetanic facilitation. Single fibre electromyography will detect the neuromuscular changes before conventional nerve stimulation; there is increased jitter and blocking which is reduced by increasing the nerve firing rate.[15] The pattern of signs and symptoms following a deliberate mass inhalation release is not known but botulinum toxin must be considered along with the nerve agents in any cases presenting with sudden cholinergic features. Pulmonary oedemagens Phosgene exposure: a model for toxic pulmonary oedema Phosgene attacks primarily attack the lower airways and alveoli as a result of limited water solubility There is a primary covalent attack on NH2 and SH substrates with free radical releases Potential cell targets are type I and II pneumocytes and alveolar macrophages. Platelet aggregation in capillaries also occurs. Later there is a second line of attack with the release of prostaglandins causing vasoconstriction, vasodilatation and bradykinin causing increased capillery permeability In addition release of 5 HT causes constriction of postcapillary vessels Thromboxane A2 leads to vasoconstriction and complement activating enzymes cause leucotriene release.[18] The use of cortocosteroids in toxic PE Theoretically there should be many advantages such as the inhibition of phospholipase A2 via induced lipomodulin and macrocortin. inhibition of macrophages, inhibition of production of prostanoids and leucotrienes and stimulation of surfactant production in type II cells. However Diller[18] felt the case for use to be unproven. Beneficial effects only follow early administration while late administration may be deleterious (inhibition of production of type I cells and enhanced fibroblast production). More recently studies have been published on animal models. Demnati et al[19] showed a reduction in airway resistance following the adminstration of dexamethasone to chlorine exposed. Gunarsson and Walther[20] published results from a pig study where inhaled beclomethasone showed improved PaO2, improved V/Q and less histological damage. However species differences exist and results should be applied with caution to man. At the present time use of inhaled steroids for developing toxic PE in man should not be discounted given the limited therapeutic options. Another tool to combat the development of toxic PE is the use of acetyl cysteine. The rationale is to increase intracellular glutathione levels to prevent lipid peroxidation induced PE. It is known that phosgene reacts with-SH groups and reduces GSH levels. Sciuto et al[21] have published a study in which phosgene-exposed rabbits receiving n-acetyl systeine treatment produced lower pulmonary wet weights and higher GSH levels inhibiting production of inflammatory leucotrienes Ventilation and developing toxic PE Hydrogen cyanide In the body HCN is broken down by rhodanase which detoxifies cyanide to thiocyanate. This process can be accelerated by provision of sodium thiosulfate which provides a store of sulfane sulfur for the enzyme.[24] The usual dose of sodium thiosufate is 50 ml 0f 25% solution. (pediatric dose 1.65ml/kg of 50% solution) It is usually administered in conjunction with sodium nitrite (300 mg given iv over 10 minutes (pediatric dose 0.15-0.33 ml/kg of 3 % solution) which causes the formation of methaemoglobin. This acts as a scavenger for HCN reducing plasma levels. Sympathomimetic support may be required for hypotension produced by sodium nitrite. HCN also reacts with heavy metals and this is the basis of the use of dicobalt edetate and hydroxycobalamin as cobalt providers for this reaction. Cobalt ions themselves are toxic but the toxicity can be countered by giving glucose which is part of standard therapy Dicobalt edetate is thought to be more effective in the binding of cyanide ions that methemoglobin despite its secondary effects of hypertension and nausea. It has long been believed that oxygen therapy and ventilation has no role in the treatement of cyanide intoxication since the blood is fully oxygenated in this condition. This view has however recently been challenged since some studies have shown that oxygen enhances the antidotal effects of the classical cyanide antidotes. Toxic agents causing direct cellular disruption (vesicants and toxins) Sulphur mustard (bis-2-chloroethyl sufide) is a colorless or pale yellow oily liquid which indeed smells faintly of mustard. Its odour threshold is 1.3 mg /m3 which is below the concentrations usually reached in a battlefield and thus several minutes of detection are possible before incapacitating doses are reached. The LCt50 is about 1500mg /min/m3 and the LCt 50 between 200 and 1000 mg/min/m3. Although the latency of action in cooler climates is about 4 hours information from the Iran-Iraq War, the scene of its most recent use, indicates that at higher ambient temperatures the agent has a far shorter latency and causes significant respiratory damage apart from its classical action as a skin vesicant.[29] Following exposure to mustard gas there is a latent period of between 4-12 hours following which there are occular symptoms comprising eye pain, blurred vision and lachrymation accompanied by a diffuse erythema of exposed skin with oedema and first degree burns. The groin and genital area are particularly susceptible. Exposure to high doses produces severe cutaneous injury with necrosis. The burns bear some resemblance to thermal burns but are very slow to heal and are prone to secondary infection. The bullae characteristic of exposure to mustard agent are filled with a fluid that is not itself corrosive. A feature of exposure is that fluid-filled bullae appear for several days afterwards in an apparently random way (i.e. not cropped). The respiratory effects of mustard exposure are important, particularly at high ambient temperatures. Following exposure there is an early tracheobronchitis with dry cough and hoarseness Heavy exposure will produce severe damage to the tracheal and main bronchial architecture with necrosis, sloughing and blockage A chemical bronchiolitis occurs at lower doses in high ambient temperatures and causes severe bronchospasm, requiring ventilation and intensive care. Lung damage following mustard exposure can be severe and permanent with chronic obstructive airways disease, bronchiectasis and reactive airways dysfunction syndrome. Mustard agent acts at a cellular level forming highly reactive sulfonium ions which attack DNA by alkylation of sulfhydryl and and amino groups. This causes the epithelial manifestations of exposure and also long term carcinogenesis, particularly of the skin, pharynx and respiratory tract. There is no specific treatment of mustard agent exposure but experimental studies in animals have shown a combination of sodium thiosulfate vitamin E and dexamethasone can improve survival and reduce organ damage[30] Willems has reviewed information concerning the clinical management of mustard gas casualties. The need for a proactive approach to airway management and ventilation is evident and intubation should be done early to allow adequate ventilation and access for debridement of the large airways. Willems[27] reported that 87% of patients requiring ventilation died and thus the onset of severe respiratory symptoms is a serious development. Another point of concern for management is the leukopenia that follows exposure to mustard gas. This becomes evident 3-5 days after exposure and usually reaches its lowest point 7-9 days after exposure. Cellular replacement either peripherally or as marrow could be considered here since mustard is bound very quickly in the body following exposure and will not cause destruction of new cells. Ricin Conclusions
References
Copyright 2003 - Indian Journal of Critical Care Medicine Free full text also available from: http://www.ijccm.org/article.asp?issn=0972-5229;year=2003;volume=7;issue=4;spage=250;epage=256;aulast=Baker The following images related to this document are available:Photo images[cm03004b4.jpg] [cm03004b3.jpg] [cm03004b2.jpg] [cm03004b1.jpg] [cm03004b5.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}