
|
Indian Journal of Critical Care Medicine
Medknow Publications on behalf of the Indian Society of Critical Care Medicine
ISSN: 0972-5229 EISSN: 1998-359x
Vol. 9, Num. 1, 2005, pp. 52-63
|
Indian Journal of Critical Care Medicine, Vol. 9, No. 1, January-March, 2005, pp. 52-63
Review Article
Recent trends in the management of status epilepticus
Rajshekher G
Consultant Neurologist, Suite No. 16, A-Wing, Apollo Hospitals, Jubilee Hills, Hyderabad - 5000033, India
Correspondence Address: Dr. G. Rajeshekher, Consultant Neurologist, Suite No. 16, A-Wing, Apollo Hospitals, Jubilee
Hills, Hyderabad - 5000033, India, E-mail: drphaniraj@apollohospital.net
Code Number: cm05010
Abstract
Status Epilepticus (SE) is a neurologic emergency associated with high morbidity and mortality. The etiology varies among the different age groups, and it has a U-shaped incidence curve, being more common at the extremes of ages. Mortality is rarely due to the status itself, and the outcome depends to a great extent on the underlying etiology and the presence of additional medical conditions. Outcome also depends on the rapidity of diagnosis and initiation of appropriate therapy. Anti-epileptic drug administration in appropriate doses should begin promptly after the suspicion of SE; intra-muscular midazolam and rectal diazepam administered by paramedical staff involved in transporting the patient also has been shown to shorten the duration of SE. Attention should be paid in the initial stages itself to airway patency, adequacy of breathing and ventilation, the circulatory status, securing intravenous access and identifying the underlying cause. The goals of therapy include rapid termination of clinical and electrical ictal activity, prevention of aspiration pneumonia, and treatment of complications in anticipation. Every hospital needs to manage SE on the basis of established protocols, and an early decision regarding artificial ventilation and midazolam or barbiturate anesthesia for refractory SE needs to be taken. With the existing protocols and available drugs, it is generally possible to control seizures and prevent complications and mortality.
Keywords: Status Epilepticus (SE), Refractory SE, Non-convulsive
SE (NCSE), lorazepam, midazolam, barbiturate anesthesia
SE is a medical
emergency in which the patient has continuous or rapidly repeating seizures.[1] The
ILAE defined SE as a seizure that ".persists for a sufficient length of time or is repeated frequently enough that recovery between attacks does not occur".[2] This
definition is a little difficult to use in clinical practice due to the
lack of specific duration, and more recent publications have tried to
remedy this by defining SE as a seizure that lasts for 20-30 minutes.[3],[4] This
time frame is based on an estimate of duration necessary to cause damage
to cerebral neurons.[3],[4] However
the practical experience of clinicians has been that seizures need to
be controlled much earlier than 20 minutes, and therefore, an operational
definition for SE has been proposed: either continuous seizures lasting
at least five minutes or two or more discrete seizures between which
there is incomplete recovery of consciousness.[5],[6]
Clinical features SE may be of convulsive type, when motor activity in the form of tonic, tonic-clonic, or clonic limb-jerking may be obvious to the observer. Patients are usually unresponsive during this stage if the seizures are generalized. Later on, the motor activity may become subtle, and may be missed, especially if the patient is seen for the first time in this phase. This is the non-convulsive phase of SE, and only upon careful observation one may notice small-amplitude twitching movements of the face, hands, or feet or nystagmoid jerking of the eyes.[7] Sometimes, even these subtle manifestations may not be visible, and the diagnosis of ongoing seizure activity depends on electroencephalography.[8] Electrographic seizures of this type are seen predominantly in the hospitalized, comatose patient, and may be more common than previously thought.[9] In a study of 236 patients with coma and no seizure activity monitored with EEG as part of coma evaluation, 8% met the criteria for non-convulsive SE (NCSE).[10] Electrographic SE or NCSE is mistakenly thought by many to be a benign condition; these patients are equally at risk for brain damage,[11],[12] even if they have been paralyzed and sedated for respiratory management. NCSE in adult animals too leads to widespread neuronal necrosis in vulnerable regions, although lesions develop more slowly than they would in the presence of convulsions or anoxia.[13] Incidence and prevalence of SE SE is a common neurologic emergency; data emanating from the Rochester Epidemiology Project (Rochester, Minnesota, USA)[14] indicate that the age-adjusted incidence of SE was 18.3 per 100,000 population in the decades 1965-84. It has a U-shaped incidence curve, peaking under 1 year and over 60 years of age.[14],[15],[16] Cumulative incidence is 4 per 1,000 to age 75 and shows the greatest increase after 60 years of age.[14] The incidence of SE is greater among males, in seizures related to acute symptomatic etiology, and for partial SE (than generalized SE). Based on the incidence of SE actually determined in Richmond, Virginia, it was projected that each year there would be 1, 26, 000 to 1,95,000 events of SE in the US, with 22,200 to 42,000 of these ending in death.[17] Statistics for Indian population are scarce; 16 (3%) of 2531 patients followed over a 3-year period in a single center (Nizam′s Institute of Medical Sciences, Hyderabad) developed SE.[18] However, this is a gross underestimate of the incidence of SE, and a study involving multiple centers alone would give a correct estimate of the actual incidence.
Causes of SE The single-most common cause of SE is non-compliance with AED drug prescription;[19] of 98 patients over the age of 14 years, this accounted for the status in 53% of patients with previous seizures.[19] Other patient-related factors include incomplete drug absorption, vomiting, and drug-interactions.[19] Other causes, especially in those without a past history of seizures, include alcohol-withdrawal, cerebrovascular disease, cerebral tumors or trauma (involving especially the frontal lobe), intracranial infection, metabolic disorders, drug overdose and cardiac arrest.[19],[20],[21] Among Indian patients, infections of CNS and neurocysticercosis together account for 62.5% of the etiological factors for SE.[18],[22] The etiology of the status is frequently multi-factorial, and no specific cause can be found in 15 % of patients.[19] In the San Francisco General Hospital, 154 patients were treated for SE in the decade between 1980 and 1989;[23] of these 154 patients reviewed, the four leading etiologies for SE were anticonvulsant drug withdrawal (39), alcohol-related (39), drug toxicity (14), and CNS infection (12).[23] Outcome in SE The most important factor deciding outcome in SE is the underlying etiology.[24] In the San Francisco study, the best response to anticonvulsants occurred in patients with SE related to tumor, anticonvulsant drug withdrawal, or refractory epilepsy.[23] The poor responders in this study had anoxia, drug toxicity, CNS infection, or other metabolic abnormalities. Aminoff et al found that the outcome of SE worsened with an increase in the duration of status.[19] Towne et al also found that the group with SE lasting <1 h had a lower mortality as compared with seizure duration one hour or more.[20] Hyperthermia, peripheral leucocytosis and CSF pleocytosis are common accompaniments of SE, and even systemic acidosis may occur in a few patients, without necessarily portending a worse outcome.[19] The overall mortality among adults with SE is 20%, and those with first-time episodes of SE are at substantial risk for more such episodes and for the development of chronic epilepsy.[17] When SE is related to an acute medical problem such as renal failure, sepsis, electrolyte abnormality, CNS infections, stroke, head trauma, drug toxicity, or hypoxia, seizures are especially difficult to control and associated with a higher mortality.[20],[23],[25] The best example of this is the elderly patient who has survived an episode of cerebral hypoxia, and has developed myoclonic SE, which carries a grave prognosis. In a recent study evaluating variables affecting outcome in children with SE, no deaths were due to SE itself;[26] symptomatic etiology (acute or remote) and refractory SE were associated with adverse outcomes, and age <12 months at development of SE, and duration of SE >60 minutes tended to be more frequent among those who developed adverse outcome.[26] Pathophysiology
Isolated seizures occur due to the generation and spread of abnormal electrical activity among neuronal networks; the networks are probably abnormal to start with. But several mechanisms come into play with the onset of a seizure, which work to terminate the attack. SE is believed to be due to the failure of these seizure-abortive mechanisms.[1]
In late 1987, following the consumption of mussels contaminated with domoic acid in Canada, there was an outbreak of gastrointestinal and neurological abnormalities;[27] the latter included seizures, leading in a few cases to SE, and 4 of these who died underwent autopsy, which showed neuronal necrosis and loss, predominantly in the hippocampus and amygdala, in a pattern similar to that observed in the kainate model of epilepsy. Both Domoic acid and kainic acid are homologues of the excitatory amino acid glutamic acid. From these findings followed the hypothesis that SE occurs due to abnormally persistent excessive excitation.[27] The other idea that there is ineffective recruitment of the inhibitory mechanisms springs from the observation of SE with penicillin and related compounds that antagonize the principal inhibitory amino acid in the brain, GABA.[28] It is now believed that loss of GABA-mediated inhibitory synaptic transmission in the hippocampus is critical for the emergence of SE, and excitatory synaptic transmission is important in sustaining SE.[29] Experimental studies in rats have shown that the sensitivity of GABA-A receptor to benzodiazepines, and other allosteric modulators decreases over time as SE continues.[29],[30] This may be one of the reasons for the failure of the inhibitory mechanisms.
Prolonged seizures produce CNS damage.[31] The
physiologic consequences of SE, such as elevation of body temperature,
transient metabolic acidosis, and elevation of hormonal concentrations
(such as epinephrine in the arrhythmic range) add to the injury.[32]
Marked rise in pressure in the systemic as well as the pulmonary circulation
may have deleterious effects, such as by causing pulmonary edema.[31],[33] Prolonged
and repeated seizures themselves cause damage to limbic structures
like the hippocampus.[34] This
damage is partly due to glutamate-mediated excitotoxicity, and not
merely because of increased metabolic demands of repetitive neuronal
firing. Continued epileptic activity may lead to relative cerebral
hypoxia and hypoglycemia.[35] The
seizures compromise cerebral vascular auto-regulation, which in turn
compromises hypothalamic autonomic regulation, and intra-cranial hypertension
may then supervene.[36] Complications
such as cardiovascular collapse, arrhythmias, aspiration pneumonia,
acute lung injury, and pulmonary hypertension may further compromise
cerebral oxygen delivery. Cerebral and systemic hypoxia and acidosis,
hyperthermia, rhabdomyolysis, and DIC may then lead to multiple organ
failure and death.
Management
The management of the patient with SE should normally occur at two levels:
- Management of the seizures themselves
- General medical management
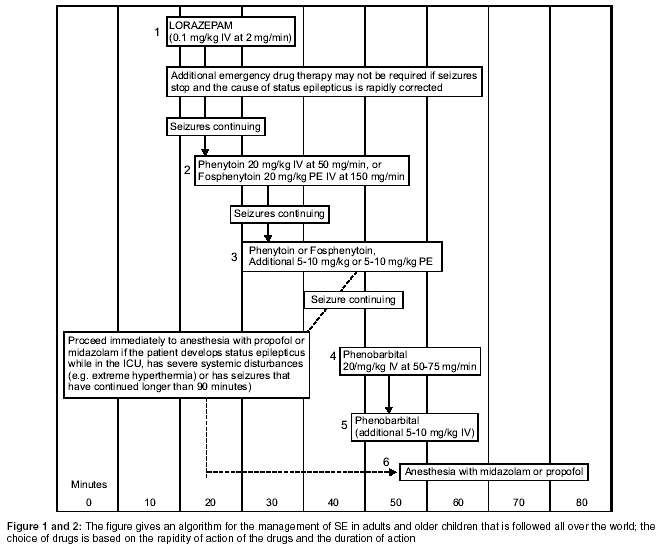
[Figure - 1] provides the algorithm for the general management of SE
presenting to the Emergency Room.
The patient should be placed in a position to minimize trauma to convulsing
limbs from neighboring hard objects and surfaces. Frequent oro-pharyngeal
suctioning may be required. Attempts to prevent tongue bites by placing
handkerchiefs and other objects in the mouth between the teeth, have led
to choking and death; a towel, folded cylindrically and placed sideways
in the mouth may be far safer.
The immediate assessment of breathing and securing the airway is of
paramount importance in the actively convulsing patients, as they are
highly prone
to develop aspiration and all the attendant complications. During the tonic-clonic
phase of the convulsion, the patient may stop breathing and become cyanosed;
however this is generally short-lasting, and does not create a problem,
unless the airway is blocked. Administration of 100% oxygen is usually
sufficient, but the airway patency should be secured with an oral or nasopharyngeal
airway tube. If there is respiratory compromise, an emergent intubation
may be called for; if neuromuscular blockade is deemed necessary to perform
the procedure, then a short-acting agent like 0.1mg/kg of vecuronium should
be employed, thereby ensuring that ongoing seizures are not missed by the
attending physician.
Alcohol ingestion is a common cause of presentation in the emergency
with SE;[37] prompt
administration of thiamine is therefore essential, often before it can
be ascertained that alcohol has been consumed. Similarly, immediate blood
sugar measurement is now routine, and even when initially normal, 100-200
ml of 25% glucose are administered in actively convulsing patients,
as blood sugar levels tend to fall, and hypoglycemia can add to the complications.
Hyperthermia and metabolic acidosis occur relatively frequently in
SE; together with the peripheral blood leucocytosis, they may suggest
an infection
and lead to the inappropriate use of antibiotics. Later on the patient
may actually develop aspiration pneumonia, for which antibiotics may
become necessary. On the other hand, the classical symptoms and signs
of acute
bacterial meningitis may be absent in convulsive SE with fever; a high
index of suspicion for acute bacterial meningitis is therefore of paramount
importance. The most appropriate management is early parenteral antibiotics
and lumbar puncture if there are no contraindications.[38] Metabolic
acidosis gets corrected once the seizures are controlled. Hyperthermia
should be managed emergently with anti-pyretics and cooling blankets,
as continued high fever can have deleterious effects on the central nervous
system.
Control of seizures: Drug therapy
In the management of SE, the response to treatment and the ultimate outcome are very much dependent on the duration of the status before effective anti-epileptic medications are administered. This was amply demonstrated in the study in San Francisco in the 1980s,[23] where
the authors found that if the AED was administered within 30 minutes of
the onset of SE, the response rate was as high as 80%, whereas in those treated after the 30 minute period, the response rate fell to 40%.
Rapid functional plasticity of GABAA receptors has been demonstrated to
occur during SE in rats with a substantial reduction of diazepam potency
for termination of the seizures,[29] especially as the duration of electrographic seizures increases.[39]
The
ideal drug to control status should be easy to administer, should produce
the effect immediately, have long-lasting effect, and at the same time
should not depress cardio-respiratory function or the consciousness. Benzodiazepines
like diazepam and barbiturates carry the risk of respiratory depression
and also depress consciousness in a dose-dependant manner. Phenytoin and
fosphenytoin can cause hypotension and cardiac arrhythmias if administered
too fast; this can be a limiting factor when attempting rapid seizure control,
though in practice this is seldom the reason for failing to control seizures.
The [Figure - 2] gives an algorithm for the management of SE in adults
and older children that is followed all over the world; the choice of drugs
is based on the rapidity of action of the drugs and the duration of action.
Lorazepam has an extremely rapid onset of action; in a retrospective analysis
comparing the treatment of SE with diazepam and lorazepam, both were found
to be equally effective, but there were fewer recurrences with lorazepam,
and fewer repeat doses were required.[40] Based on this the authors recommended that lorazepam should be the drug of first choice. Phenytoin has the advantages of the availability of an injectable preparation, and till recently was the only other anti-epileptic drug whose plasma levels could be rapidly brought to the therapeutic range. In addition it has a long duration of action. In a five-year randomized, double-blind trial comparing the efficacy of lorazepam alone, phenytoin alone, diazepam with phenytoin, and phenobarbital alone for the treatment of generalized SE, the treatments were equally effective, except that lorazepam alone was more effective than phenytoin alone, when seizures were assessed 20 minutes after the administration began.[41] In the algorithm, lorazepam is followed by Phenytoin if the seizures are not controlled, and this is preferred by neurologists and epileptologists the world over. When the cause of the SE is a reversible one, such as sub-therapeutic drug concentration or an acute metabolic process, then lorazepam alone may be sufficient and obviate the need for Phenytoin or Fosphenytoin.
Pharmacologic Therapy
Benzodiazepines
The benzodiazepines including diazepam, lorazepam, and midazolam are the preferred initial choice for therapy of SE, mainly because of their rapidity of onset of action, and high efficacy in aborting seizures; benzodiazepine-receptor-mediated enhancement of GABAergic transmission is their primary mode of action. In addition, they also block sustained repetitive neuronal firing in a manner similar to carbamazepine and phenytoin. All benzodiazepines rapidly enter CSF, with peak concentrations usually attained within 15 min of dosage.[42] Though lorazepam is less lipid-soluble, and therefore its serum and CSF levels rise much more slowly than that of diazepam, it remains longer in brain than in the serum, leading to increasing brain: serum ratios over time.[43] In a randomized, double-blind trial comparing standard doses of diazepam and lorazepam, both were found to be equally effective, the median time to termination of seizure also being similar (two minutes for diazepam, three for lorazepam).[44] Despite these similarities, the important difference between diazepam and lorazepam is their duration of actions (12 to 24 hours for lorazepam versus 15 to 20 minutes for diazepam), and it is this property which makes lorazepam superior in the acute treatment of SE.[45] Adverse
effects occur in 13% of the lorazepam-treated patients and in 12% of
the diazepam-treated patients.[44] These
include respiratory depression in 3 to 10%, hypotension in <2%, and impaired consciousness in 20 to 60% patients.[44],[46],[47] One problem with continued use of benzodiazepine in SE is that prolonged seizures of SE rapidly alter the functional properties of hippocampal dentate granule cell GABA(A) receptors leading to loss of sensitivity of the animals to diazepam during SE.[29] Since this is not seen with barbiturates, they are preferred for refractory SE.
Phenytoin and Fosphenytoin
Phenytoin is ideally suited for rapid control of seizure as well as
for prolonged effect, so much so that frequently, epilepsy treatment is
equated with the administration of phenytoin. Even though many more alternatives
are now available, phenytoin still has many advantages not offered by the
other medications. Its use in the acute setting is most useful in three
situations: (1) after rapid control of seizure with benzodiazepines, for
prolonged effect; (2) as initial therapy for SE, and (3) when benzodiazepines
fail to control seizures. The most common problem with its use in the emergency
department is inadequate dose; at least 20 mg/kg is required, and an additional
5-10 mg/kg may occasionally be required.[48] The
maximum tolerated rate of administration is 50 mg/min; given at this rate
it causes hypotension in 28-30% of patients, and cardiac arrhythmias (bradycardia or ectopic beats) in 2%,
especially in those above 50 years age and those with pre-existing cardiac
disease.[49] These cardiovascular
side-effects are attributed to phenytoin itself and to its propylene glycol
diluent, and can be mitigated by slowing the rate of administration.[50] This
means that the shortest time in which a 1000 mg dose can be administered
is 20 minutes; in practice, it is administered even more slowly, and this
is occasionally the reason for its failure. In a known epileptic who is
already on phenytoin, presenting with SE, a serum sample for estimation
of phenytoin levels should be drawn prior to administration of anti-epileptics;
in this setting, the serum phenytoin level is likely to be sub-therapeutic
but not zero, and that is why it is common practice to administer a half-loading
dose of phenytoin. Though one should not wait for the serum levels to treat
SE, this practice (of administering a half-loading dose) is at best an
educated guess and the ER physician should not hesitate to administer additional
phenytoin if the seizures remain uncontrolled. When a typical loading dose
is given to an adult, it takes about 20-25 minutes for maximal effect.[50] Fosphenytoin
is the disodium phosphate ester pro-drug of phenytoin for parenteral use.[51] It
has significantly better water solubility, but little intrinsic pharmacologic
activity. After parenteral administration, it is rapidly and completely
converted by serum and tissue alkaline phosphatases to free phenytoin.
The conversion half-life is approximately 8 to 15 minutes. Fosphenytoin
has been recommended over conventional phenytoin for SE mainly for three
reasons;[52] (1) It has a
pH of 8.5 as against 12 for phenytoin, (2) The vehicle does not contain
propylene glycol or ethanol, thereby reducing the potential for cardiovascular
or cutaneous side-effects, and (3) It can be administered much faster than
phenytoin; Intravenous fosphenytoin is tolerated at infusion rates up to
three times faster than those for phenytoin, and therapeutic concentrations
are established within 10 minutes.[53],[54],[55],[56] No
clinically significant hypotension or cardio-vascular side-effects have
been reported with fosphenytoin, and injection-site-related problems like
phlebitis and soft-tissue damage are less common with fosphenytoin. Another
potential advantage of substituting fosphenytoin for phenytoin is the prevention
of the "purple glove syndrome", which is reported in 6% of patients
receiving intravenous phenytoin administration, characterized by progressive
distal limb edema, discoloration, and pain.[57]
Phenobarbital
Phenobarbital, 20 mg per kilogram at a rate of 50 to 75 mg per minute,
has been used in the past when benzodiazepine and phenytoin fail; in a
small, randomized, non-blinded trial involving 36 patients, phenobarbital
was found to have shorter cumulative convulsion time and response latency
when compared to benzodiazepine and phenytoin combined.[58] However,
Phenobarbital depresses respiratory drive, level of consciousness, and
blood pressure, all effects are accentuated when it follows the administration
of benzodiazepine.[59]
Treatment of Refractory SE
When SE does not respond to standard treatment with benzodiazepine,
phenytoin, and phenobarbital, then it is considered to be refractory, requiring
aggressive management. Patients generally need to be in the intensive care
unit, and most require intubation to prevent aspiration and mechanical
ventilation. This therapy requires a team approach, with the anesthetist
and intensivist playing vital roles. Infusions with anesthetic doses of
midazolam or propofol are usually required. EEG monitoring is generally
necessary, and the aim is suppression of epileptic spikes; the end-point
is burst-suppression, though occasionally, fall in BP becomes a limiting
factor, especially when propofol is used. Midazolam is given in a bolus
dose of 0.2 mg/kg slow intravenous push, followed by 0.75 to10 µg/kg
per minute.[60],[61] Alternatively,
propofol at a dose of 1 to 2 mg per kilogram intravenous followed by 2
to 10 mg/kg/hr may be used.[62] It
directly activates GABAA receptors. In addition, propofol inhibits the
NMDA receptor and modulates calcium influx through slow calcium ion channels.[63] Propofol
has a rapid onset of action with a dose-related hypnotic effect, and recovery
is rapid even after prolonged use. Propofol decreases cerebral oxygen consumption,
reduces intracranial pressure and has potent anti-convulsant properties.
It is a potent antioxidant, has anti-inflammatory properties and is a bronchodilator.
As a consequence of these properties propofol is being increasingly used
in the management of traumatic head injury, SE, delirium tremens, status
asthmaticus and in critically ill septic patients.[63] Propofol
has a remarkable safety profile: dose dependent hypotension is the commonest
complication, particularly in patients who are volume-depleted, or have
limited cardiac reserve.[64] Hypertriglyceridemia
and pancreatitis are uncommon complications.[63] High
dose propofol infusions have been associated with the "propofol syndrome";
this is a potentially fatal complication characterized by severe metabolic
acidosis and circulatory collapse.[63] A
smaller proportion of patients respond to propofol, than to barbiturates,
but the response appears much earlier (2.6 min versus 123 min with barbiturate
coma).[65],[66] The
plasma concentrations of propofol associated with control of SE were 14 µM ± 4 (2.5 µg/ml).
Recurrent seizures are common when propofol infusions are suddenly discontinued
but not when the infusions are gradually tapered.[67] Continuous
electroencephalographic monitoring is necessary. The duration of such treatment
is never certain, but usually, a seizure-free period of about 24 hours
is sufficient, and the agent can be gradually tapered thereafter, unless
further seizures supervene. Recovery usually takes a long time, depending
on the duration of infusion, occasionally taking as long as 36-72 hours.
More recently a meta-analysis of 22 studies with original data on the use
of propofol in SE,[68] has
raised serious doubts about the safety of propofol in refractory SE, because
two non-randomized studies and several case reports show an increased risk
of mortality. The authors of this meta-analysis advise that guidelines
should not recommend the use of propofol as a routine treatment in refractory
SE before a proper randomized trial has been performed. Midazolam, on the
other hand, appears safer; in a recent report of the treatment of 27 pediatric
patients with refractory generalized convulsive SE, midazolam infusion
at a rate of 3.1 µg/kg/min was found to be effective and safe in
26, without adverse effects such as hypotension, bradycardia or respiratory
depression.[69] In a randomized
open-label study in children at the PGIMER, Chandigarh, comparing the efficacy
of midazolam and diazepam in refractory SE, both were found to be equally
effective, with median time to seizure control of 16 minutes; however in
the midazolam group, seizures recurred in more children (57% versus 16% in
diazepam group; P <0.05) and the mortality was higher in the midazolam group (38%) as compared to the diazepam group (10.5%, P <0.1>0.05).[70] The
maximum dose (mean ± SD) of midazolam and diazepam required was 5.3 ± 2.6 µg/kg/min and 0.04 ± 0.02
mg/kg/min, respectively. Thus the experience with midazolam also is varied
and mixed, though overall it appears to be equally effective and marginally
safer.
Benzodiazepines and barbiturates enhance GABAA receptor-mediated inhibition.
However, patients often become refractory to benzodiazepines when seizures
are prolonged, and barbiturates are often then used for these refractory
cases of SE.[29] RSE has been
treated conventionally with high-dose intravenous barbiturate coma;[71] pentobarbital
coma (PBC) was evaluated in a small series of 17 patients with RSE.[72] Seizures
were aborted in 16 of 17 patients, but vasopressors were required in 11
for severe hypotension; nine of them died, and among these, new-onset epilepsy,
multiorgan failure before or during PBC, age >40 years, and hypotension requiring vasopressors during PBC were the causes identified. In a meta-analysis of twenty-eight studies describing a total of 193 patients comparing the efficacy of midazolam (MDL), propofol (PRO), and pentobarbital (PTB) for terminating seizures and improving outcome in RSE patients, PTB treatment was associated with a lower frequency of short-term treatment failure (8 vs. 23%; P <0.01), breakthrough seizures (12 vs. 42%; P <0.001), and changes to a different AED (3 vs. 21%; P <0.001) and a higher frequency of hypotension (systolic blood pressure <100 mm Hg; 77 vs. 34%; P <0.001).[73] The
conclusion is that though PB is more effective, midazolam is both safe
and effective.
Sodium Valproate
Although sodium valproate is not approved by the US FDA for treatment
of SE, it was found to have an overall efficacy of 63.3% in a study
in which 63 patients were given a median dose of 31.5 mg/kg of IV valproate.[74] In
a multicenter, open-label, prospective, dose-escalation study of IV sodium
valproate administered to patients with epilepsy, rates of infusion of
up to 6 mg/kg/minute and doses of up to 30 mg/kg were well tolerated, with
no clinically significant negative effects on blood pressure and pulse
rate and caused only mild-to-moderate, reversible adverse events,[75] even
among unstable SE patients with hypotension.[76]
Topiramate
Topiramate is an anticonvulsant with multiple activities at receptors
and ion channels that may be more effective than conventional anticonvulsants
in treating RSE. Like phenytoin, topiramate exhibits voltage-sensitive,
use-dependent, sodium-channel blockade and may have an additive effect
at this site.[77] Topiramate
potentiates GABA inhibition independently of the benzodiazepine site on
the GABAA receptor and significantly elevates brain GABA levels; this more
likely underlies its effectiveness in RSE.[78],[79] Another
action of topiramate is its ability to antagonize excitatory glutamatergic
transmission, providing a mechanism for termination of seizure discharges
in RSE.[80] Topiramate has
been shown to reduce neuronal injury after prolonged SE and may prevent
delayed neuronal death.[81] In
a series of six cases, topiramate effectively terminated RSE in a variety
of clinical settings.[82] In
cases of RSE unresponsive to sequential trials of multiple agents, a suspension
of topiramate administered via nasogastric tube was effective in aborting
RSE; effective dosages range from 300 to 1,600 mg/d. Except for lethargy,
no adverse events were reported.[82]
Surgery for SE
Patients with RSE of focal origin may be potentially amenable to resective
surgery.[83] The literature
is limited to isolated case reports or small case series involving multiple
subpial transections, cortical resection, corpus callosotomy, or implantation
of a vagus nerve stimulator.[84],[85] At
the Jefferson Comprehensive Epilepsy Center, patients with medically intractable
SE who fail to respond to three courses of cerebral suppressant therapy
for approximately 2 weeks are considered for surgical treatment in the
absence of any known remitting etiology. When structural or functional
neuroimaging shows a focal lesion, or the EEG displays focal changes, they
prefer focal resection and/or subpial transection. Corpus callosotomy is
used for patients with generalized or non-localizable intractable SE.[85] Bingaman
and colleagues at the Cleveland Clinic performed resective surgery in the
acute setting for refractory SE in 10 patients with focal epileptogenesis
when High Dose Suppressive Therapy (HDST) failed; 7 (10%) became seizure-free, and 3 (30%)
had significant improvement in epilepsy.[84]
Pre-hospital treatment
SE frequently occurs or is identified in settings where it may not
be feasible to administer intravenous drugs; in these settings, rectal
diazepam, especially in children, and intramuscular midazolam can be used.
Rectal diazepam is very easy to administer: a starting dose of 0.5 mg/kg
is recommended, with a maximum of 20 mg per single dose;[86] this
is effective in 67% within 15 minutes. A gel formulation for rectal
administration of diazepam is under study for rapid out-of-hospital control
of SE.[87] After intramuscular
administration midazolam is rapidly absorbed (mean time to peak serum concentrations
25 min),[88] and seizures
are controlled within 10 minutes.[89]
The mean absolute bioavailability of intramuscular midazolam is 87% and
after intramuscular administration, sedation is slower (2-30 min versus
less than 1-2 min with the intravenous preparation) and persists for a
longer period (20-120 min versus 7-75 min).[88]
Diazepam is administered to children in SE by paramedics in many Emergency
Medical Services systems throughout the United States despite the lack
of clear evidence that this therapy is safe and effective when employed
in the pre-hospital environment. In a retrospective review, published in
1995,[90] pre-hospital diazepam
therapy was associated with SE of shorter duration (32 min vs. 60 min; P =0.007)
and a reduced likelihood of recurrent seizures in the emergency department
(58% vs. 85%; P =0.045). Though these data suggest
that pre-hospital administration of diazepam may shorten the duration of
SE in children and simplify the subsequent management of these patients
in the emergency department, data concerning the safety of such treatment
are scanty. Possibly, rectal diazepam or intramuscular midazolam may be
considered relatively safe and effective in this setting.
Treatment after recovery from SE
Recovery in the post-SE phase is mainly dependent on the underlying
cause of the SE, other medical problems, and complications related to
the episode of SE, such as aspiration pneumonia. At this stage, the patient
is likely to be on multiple AEDs, and this may be partly responsible
for
the morbidity. The aim at this stage is to minimize the number of drugs
the patient has to take; optimizing anti-epileptic medication involves
giving the appropriate drug in the lowest effective dose to minimize
adverse effects. One problem that can arise while doing this is that
of drug interactions.
When the dose of an effective AED is decreased, and another interacting
medication, such as an antibiotic, is withdrawn, the AED dose may become
insufficient, and the patient may have breakthrough seizure. Careful
titration at this stage may involve measurement of AED serum levels.
Conclusion
SE should be identified early and treatment initiated as soon as it is
clear that a seizure has lasted 5-10 minutes; out-of-hospital administration
of intramuscular midazolam or rectal diazepam by paramedics transporting
the patient could shorten the duration of SE and hospital stay. Airway,
breathing and circulation should be assessed in the emergency department
and adequate steps initiated to correct any abnormalities. The appropriate
drug in correct doses should be administered; every hospital needs to follow
the treatment based on established protocols, and an early decision to
paralyze and ventilate the patient in preparation for continuous midazolam
or propofol administration should be taken. With the available drugs and
the facilities to manage complications, the morbidity and mortality associated
with SE can be minimized.
References
| 1. | Daniel H. Lowenstein MD, Brian K. Alldredge. Status Epilepticus. N Engl J Med 1998;338:970-6. Back to cited text no. 1 |
| 2. | Proposal for revised clinical and electroencephalographic classification of epileptic seizures: from the Commission on Classification and Terminology of the International League against Epilepsy. Epilepsia 1981;22:489-501. Back to cited text no. 2 |
| 3. | Treatment of convulsive status epilepticus: Recommendations of the Epilepsy Foundation of America's Working Group on Status Epilepticus. JAMA 1993;270:854-9. Back to cited text no. 3 |
| 4. | Shorvon S. Status epilepticus: Its clinical features and treatment in children and adults. Cambridge, England: Cambridge University Press; 1994. Back to cited text no. 4 |
| 5. | Bleck TP. Convulsive disorders: Status epilepticus. Clin Neuropharmacol 1991;14:191-8. Back to cited text no. 5 |
| 6. | Gastaut H, Broughton R. Epileptic seizures: Clinical and electrographic features, diagnosis and treatment. Springfield, Ill.: Charles CT; 1972. p. 25-90. Back to cited text no. 6 |
| 7. | Theodore WH, Porte RJ, Albert P, et al. The secondarily generalized tonic-clonic seizure: A videotape analysis Neurology 1994;44:1403-7. Back to cited text no. 7 |
| 8. | Simon RP, Aminoff MJ. Electrographic status epilepticus in fatal anoxic coma. Ann Neurol 1986;20:351-5. Back to cited text no. 8 |
| 9. | Lowenstein DH, Aminoff MJ. Clinical and EEG features of status epilepticus in comatose patients. Neurology 1992;42:100-4. Back to cited text no. 9 |
| 10. | Jordan KG. Continuous EEG ad evoked potential monitoring in the neuroscience intensive care unit. J Clin Neurophysiol 1993;10:445-75. Back to cited text no. 10 |
| 11. | Towne AR, Waterhouse EJ, Boggs JG, Garnett LK, Brown AJ, Smith JR, et al. Prevalence of non-convulsive status epilepticus in comatose patients. Neurology 2000;54:340. Back to cited text no. 11 |
| 12. | Litt B, Wityk RJ, Hertz SH, Mullen PD, Weiss H, Ryan DD, et al. Nonconvulsive status epilepticus in the critically ill elderly. Epilepsia 1998;39:1194-202. Back to cited text no. 12 |
| 13. | Brenner RP. Is it status? Epilepsia 2002;43:S103-13. Back to cited text no. 13 |
| 14. | Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia 1993;34:S37. Back to cited text no. 14 |
| 15. | Hesdorffer DC, Logroscino G, Cascino G, Annegers JF, Hauser WA. Incidence of status epilepticus in Rochester, Minnesota, 1965-1984. Neurology 1998;50:735-41. Back to cited text no. 15 |
| 16. | Aminoff MJ, Simon RP. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am J Med 1980;69:657-66. Back to cited text no. 16 |
| 17. | Chin RF, Neville BG, Scott RC. A systematic review of the epidemiology of status epilepticus. Eur J Neurol 2004;11:800-10. Back to cited text no. 17 |
| 18. | DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia; Neurology 1996;46:1029-35. Back to cited text no. 18 |
| 19. | Murthy JM, Yangala R. Acute symptomatic seizures - incidence and etiological spectrum: A hospital-based study from South India. Seizure 1999;8:162-5. Back to cited text no. 19 |
| 20. | Aminoff MJ, Simon RP. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am J Med 1980;69:657-66. Back to cited text no. 20 |
| 21. | Towne AR, Pellock JM, Ko D, DeLorenzo RJ. Determinants of mortality in status epilepticus. Epilepsia 1994;35:27-34. Back to cited text no. 21 |
| 22. | Vignatelli L, Tonon C, D'Alessandro R. Bologna Group for the Study of Status Epilepticus. Incidence and short-term prognosis of status epilepticus in adults in Bologna, Italy. Epilepsia 2003;44:964-8. Back to cited text no. 22 |
| 23. | Singhi S, Singhi P, Dass R. Status epilepticus: Emergency management. Indian J Pediatr 2003;70:S17-22. Back to cited text no. 23 |
| 24. | Lowenstein DH, Alldredge BK. Status epilepticus at an urban public hospital in the 1980s. Neurology 1993;43:483-8. Back to cited text no. 24 |
| 25. | Hui AC, Joynt GM, Li H, Wong KS. Status epilepticus in Hong Kong Chinese: Etiology, outcome and predictors of death and morbidity. Seizure 2003;12:478-82. Back to cited text no. 25 |
| 26. | Kwong KL, Chang K, Lam SY. Features predicting adverse outcomes of status epilepticus in childhood. Hong Kong Med J 2004;10:156-9. Back to cited text no. 26 |
| 27. | Teitelbaum JS, Zatorre RJ, Carpenter S, Gendron D, Evans AC, Gjedde A, et al. Neurologic sequelae of domoic acid intoxication due to the ingestion of contaminated mussels. N Engl J Med 1990;322:1781-7. Back to cited text no. 27 |
| 28. | Lothman EW, Bertram EH III, Stringer JL. Functional anatomy of hippocampal seizures. Prog Neurobiol 1991;37:1-82. Back to cited text no. 28 |
| 29. | Macdonald RL, Kapur J. Acute cellular alterations in the hippocampus after status epilepticus. Epilepsia 1999;40:S9-20; discussion S21-2. Back to cited text no. 29 |
| 30. | Kapur J, Macdonald RL. Rapid Seizure-Induced Reduction of Benzodiazepine and Zn2+ Sensitivity of Hippocampal Dentate Granule Cell GABAA Receptors. J Neurosciences 1997;17:7532-40. Back to cited text no. 30 |
| 31. | Simon RP. Physiologic consequences of status epilepticus. Epilepsia 1985;26:S58-66. Back to cited text no. 31 |
| 32. | Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus Epilepsia 1993;34:S37-53. Back to cited text no. 32 |
| 33. | Meldrum BS, Vigouroux RA, Brierley JB. Systemic factors and epileptic brain damage: Prolonged seizures in paralyzed, artificially ventilated baboons. Arch Neurol 1973;29:82-7. Back to cited text no. 33 |
| 34. | Meldrum BS, Brierley JB. Prolonged epileptic seizures in primates: Ischemic cell change and its relation to ictal physiological events. Arch Neurol 1973;28:10-7. Back to cited text no. 34 |
| 35. | Shorvon SD. Emergency treatment of status epilepticus. In: Shorvon SD, editor. Status epilepticus: Its clinical features and treatment in children and adults. Cambridge: Cambridge University Press; 1994. p. 175-292. Back to cited text no. 35 |
| 36. | Heafield MTE. Managing status epilepticus New drug offers real advantages. BMJ 2000;320:953-4. Back to cited text no. 36 |
| 37. | Hillbom M, Pieninkeroinen I, Leone M. Seizures in alcohol-dependent patients: Epidemiology, pathophysiology and management CNS. Drugs 2003;17:1013-30. Back to cited text no. 37 |
| 38. | Chin RFM, Neville BGR, Scott RC. Meningitis is a common cause of convulsive status epilepticus with fever. Arch Dis Child 2005;90:66-9. Back to cited text no. 38 |
| 39. | Walton NY, Treiman DM. Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol 1988;101:267-75. Back to cited text no. 39 |
| 40. | Cock HR, Schapira AHV. A comparison of lorazepam and diazepam as initial therapy in convulsive status epilepticus. Q J Med 2002;95:225-31. Back to cited text no. 40 |
| 41. | Treiman DM, Meyers PD, Walton NY, Collins JF, Colling CA, Rowan J, et al. For The Veterans Affairs Status Epilepticus Cooperative Study Group. A Comparison of Four Treatments for Generalized Convulsive Status Epilepticus. N Engl J Med 1998;339:792-8. Back to cited text no. 41 |
| 42. | Arendt RM, Greenblatt DJ, deJong RH, Bonin JD, Abernethy DR, Ehrenberg BL, et al. In vitro correlates of benzodiazepine cerebrospinal fluid uptake, pharmacodynamic action and peripheral distribution. J Pharmacol Exp Ther 1983;227:98-106. Back to cited text no. 42 |
| 43. | Walton NY, Treiman DM. Lorazepam treatment of experimental status epilepticus in the rat: Relevance to clinical practice. Neurology 1990;40:990-4. Back to cited text no. 43 |
| 44. | Leppik IE, Derivan AT, Homan RW, Walker J, Ramsay RE, Patrick B. Double-blind study of lorazepam and diazepam in status epilepticus. JAMA 1983;249:1452-4. Back to cited text no. 44 |
| 45. | Treatment of convulsive status epilepticus. Recommendations of the Epilepsy Foundation of America's Working Group on Status Epilepticus. JAMA 1993;270. Back to cited text no. 45 |
| 46. | Nicol CF, Tutton JC, Smith BH. Parenteral diazepam in status epilepticus. Neurology 1969;19:332-43. Back to cited text no. 46 |
| 47. | George KA, Dundee JW. Relative amnesic actions of diazepam, flunitrazepam and lorazepam in man. Br J Clin Pharmacol 1977;4:45-50. Back to cited text no. 47 |
| 48. | Osorio I, Reed RC. Treatment of refractory generalized tonic-clonic status epilepticus with pentobarbital anesthesia after high-dose phenytoin. Epilepsia 1989;30:464-71. Back to cited text no. 48 |
| 49. | Wilder BJ. Efficacy of phenytoin in treatment of status epilepticus In: Delgado-Escueta AV, Wasterlain CG, Treiman DM, Porter RJ, editors. Advances in neurology Vol 34 Status epilepticus: Mechanisms of brain damage and treatment. New York: Raven Press; 1983. p. 441-6. Back to cited text no. 49 |
| 50. | Cranford RE, Leppik IE, Patrick B, Anderson CB, Kostick B. Intravenous phenytoin: Clinical and pharmacokinetic aspects. Neurology 1978;28:874-80. Back to cited text no. 50 |
| 51. | Browne TR. Fosphenytoin (Cerebyx) Clin Neuropharmacol 1997;20:1-12. Back to cited text no. 51 |
| 52. | Koul R, Deleu D. Subtherapeutic free phenytoin levels following fosphenytoin therapy in status epilepticus. Neurology 2002;58:147-8. Back to cited text no. 52 |
| 53. | Jamerson BD, Dukes GE, Brouwer KL, Donn KH, Messenheimer JA, Powell JR. Venous irritation related to intravenous administration of phenytoin versus fosphenytoin. Pharmacotherapy 1994;14:47-52. Back to cited text no. 53 |
| 54. | Elson MA, Loewn GR, Voightman RE, Koup JR, Holmes GB, Hunt AJ, et al. Pharmacokinetics and tolerance of fosphenytoin and phenytoin administered intravenously to health subjects. Can J Neurol Sci 1993;20:S180. Back to cited text no. 54 |
| 55. | Ramsay RE, Philbrook B, Fischer JH, Sloan FB, Allen FH, Runge JW, et al. Safety and pharmacokinetics of fosphenytoin (Cerebyx) compared with Dilantin following rapid intravenous administration. Neurology 1996;46:A245. Back to cited text no. 55 |
| 56. | Ramsay RE, DeToldo J. Intravenous administration of fosphenytoin: Options for the management of seizures. Neurology 1996;46:S17-9. Back to cited text no. 56 |
| 57. | O'Brien TJ, Cascino GD, So EL, Hanna DR. Incidence and clinical consequence of the purple glove syndrome in patients receiving intravenous phenytoin. Neurology 1998;51:1034-9. Back to cited text no. 57 |
| 58. | Shaner DM, McCurdy SA, Herring MO, Gabor AJ. Treatment of status epilepticus: A prospective comparison of diazepam and phenytoin versus phenobarbital and optional phenytoin. Neurology 1988;38:202-7. Back to cited text no. 58 |
| 59. | Goldberg MA, McIntyre HB. Barbiturates in the treatment of status epilepticus. In: Delgado-Escueta AV, Wasterlain CG, Treiman DM, Porter RJ, editors. Advances in neurology Vol. 34 Status epilepticus: Mechanisms of brain damage and treatment. New York: Raven Press; 1983. p. 499-503. Back to cited text no. 59 |
| 60. | Kumar A, Bleck TP. Intravenous midazolam for the treatment of refractory status epilepticus. Crit Care Med 1992;20:483-8. Back to cited text no. 60 |
| 61. | Bleck TP. Advances in the management of refractory status epilepticus. Crit Care Med 1993;21:955-7. Back to cited text no. 61 |
| 62. | Stecker MM, Kramer TH, Raps EC, O'Meeghan R, Dulaney E, Skaar DJ. Treatment of refractory status epilepticus with propofol: Clinical and pharmacokinetic findings. Epilepsia 1998;39:18-26. Back to cited text no. 62 |
| 63. | Marik PE. Propofol: Therapeutic indications and side-effects. Curr Pharm Des 2004;10:3639-49. Back to cited text no. 63 |
| 64. | Angelini G, Ketzler JT, Coursin DB. Use of propofol and other nonbenzodiazepine sedatives in the intensive care unit. Crit Care Clin 2001;17:863-80. Back to cited text no. 64 |
| 65. | Stecker MM, Kramer TH, Raps EC, O'Meeghan R, Dulaney E, Skaar DJ. Treatment of refractory status epilepticus with propofol: Clinical and pharmacokinetic findings. Epilepsia 1998;39:18-26. Back to cited text no. 65 |
| 66. | Rossetti AO, Reichhart MD, Schaller MD, Despland PA, Bogousslavsky J. Propofol treatment of refractory status epilepticus: A study of 31 episodes. Epilepsia 2004;45:757-63. Back to cited text no. 66 |
| 67. | Stecker MM, Kramer TH, Raps EC, O'Meeghan R, Dulaney E, Skaar DJ. Treatment of refractory status epilepticus with propofol: Clinical and pharmacokinetic findings. Epilepsia 1998;39:18-26. Back to cited text no. 67 |
| 68. | Niermeijer JM, Uiterwaal CS, Van Donselaar CA. Propofol in status epilepticus: Little evidence, many dangers? J Neurol 2003;250:1237-40. Back to cited text no. 68 |
| 69. | Ozdemir D, Gulez P, Uran N, Yendur G, Kavakli T, Aydin A. Efficacy of continuous midazolam infusion and mortality in childhood refractory generalized convulsive status epilepticus. Seizure 2005;14:129-32. Back to cited text no. 69 |
| 70. | Singhi S, Murthy A, Singhi P, Jayashree M. Continuous midazolam versus diazepam infusion for refractory convulsive status epilepticus. J Child Neurol 2002;17:106-10. Back to cited text no. 70 |
| 71. | Jordan KG. Status epilepticus. A perspective from the neuroscience intensive care unit. Neurosurg Clin N Am 1994;5:671-86. Back to cited text no. 71 |
| 72. | Yaffe K, Lowenstein DH. Prognostic factors of pentobarbital therapy for refractory generalized status epilepticus. Neurology 1993;43:895-900. Back to cited text no. 72 |
| 73. | Claassen J, Hirsch LJ, Emerson RG, Mayer SA. Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: A systematic review. Epilepsia 2002;43:146-53. Back to cited text no. 73 |
| 74. | Limdi NA, Shimpi AV, Faught E, Gomez CR, Burneo JG. Efficacy of rapid IV administration of valproic acid for status epilepticus. Neurology 2005;64:353-5. Back to cited text no. 74 |
| 75. | Wheless JW, Vazquez BR, Kanner AM, Ramsay RE, Morton L, Pellock JM. Rapid infusion with valproate sodium is well tolerated in patients with epilepsy. Neurology 2004;63:1507-8. Back to cited text no. 75 |
| 76. | Sinha S, Naritoku DK. Intravenous valproate is well tolerated in unstable patients with status epilepticus. Neurology 2000;55:722-4. Back to cited text no. 76 |
| 77. | DeLorenzo RJ, Sombati S, Coulter DA. Effects of topiramate on sustained repetitive firing and spontaneous recurrent seizure discharges in cultured hippocampal neurons. Epilepsia 2000;41:S40-4. Back to cited text no. 77 |
| 78. | White HS, Brown SD, Woodhead JH, Skeen GA, Wolf HH. Topiramate modulates GABA-evoked currents in murine cortical neurons by a nonbenzodiazepine mechanism. Epilepsia 2000;41:S17-20. Back to cited text no. 78 |
| 79. | Petroff OA, Hyder F, Rothman DL, Mattson RH. Topiramate rapidly raises brain GABA in epilepsy patients. Epilepsia 2001;42:543-8. Back to cited text no. 79 |
| 80. | Gibbs JW, Sombati S, DeLorenzo RJ, Coulter DA. Cellular actions of topiramate: Blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia 2000;41:S10-6. Back to cited text no. 80 |
| 81. | Niebauer M, Gruenthal M. Topiramate reduces neuronal injury after experimental status epilepticus. Brain Res 1999;837:263-9. Back to cited text no. 81 |
| 82. | Towne AR, Garnett LK, Waterhouse EJ, Morton LD, DeLorenzo RJ. The use of topiramate in refractory status epilepticus. Neurology 2003;60:332-4. Back to cited text no. 82 |
| 83. | Alexopoulos A, Lachhwani DK, Gupta A, Kotagal P, Harrison AM, Bingaman W, et al. Resective surgery to treat refractory status epilepticus in children with focal epileptogenesis. Neurology 2005;64:567-70. Back to cited text no. 83 |
| 84. | Ma X, Liporace J, O'Connor MJ, Sperling MR. Neurosurgical treatment of medically intractable status epilepticus. Epilepsy Res 2001;46:33-82. Back to cited text no. 84 |
| 85. | Winston KR, Levisohn P, Miller BR, Freeman J. Vagal nerve stimulation for status epilepticus. Pediatr Neurosurg 2001;34:190-2. Back to cited text no. 85 |
| 86. | Hoppu K, Santavuori P. Diazepam rectal solution for home treatment of acute seizures in children. Acta Paediatr Scand 1981;70:369-72. Back to cited text no. 86 |
| 87. | Mitchell WG, Shellenberger K, Groves L, et al. Rectal diazepam gel (Diastat) for acute repetitive seizures: Results of a double-blind, placebo-controlled study in children and adults with epilepsy. Epilepsia 1996;37:154. Back to cited text no. 87 |
| 88. | Bell DM, Richards G, Dhillon S, Oxley JR, Cromarty J, Sander JW, et al. A comparative pharmacokinetic study of intravenous and intramuscular midazolam in patients with epilepsy. Epilepsy Res 1991;10:183-90. Back to cited text no. 88 |
| 89. | Lahat E, Aladjem M, Eshel G, Bistritzer T, Katz Y. Midazolam in treatment of epileptic seizures. Pediatr Neurol 1992;8:215-6. Back to cited text no. 89 |
| 90. | Alldredge BK, Wall DB, Ferriero DM. Effect of prehospital treatment on the outcome of status epilepticus in children. Pediatr Neurol 1995;12:213-6. Back to cited text no. 90 |
Copyright 2005 - Indian Journal of Critical Care Medicine
The following images related to this document are available:
Photo images
[cm05010f2.jpg]
[cm05010f1.jpg]
|

{kind=link}
{kind=link}