 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
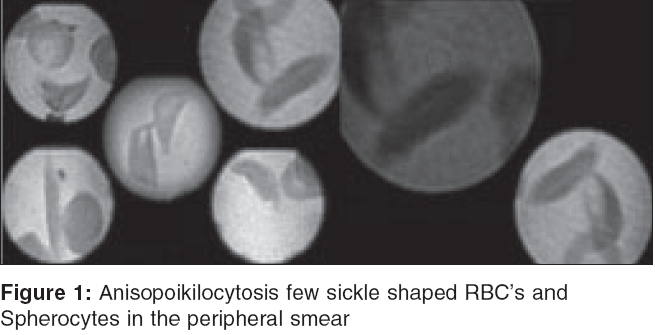
Indian Journal of Critical Care Medicine, Vol. 9, No. 2, April-June, 2005, pp. 92-95 Case Reports Sudden unexpected death in an undiagnosed sickle disease Pillai LalithaV, Husainy Saifuddin, Gosavi Sameer, Vaidya Narendra Dept. Of Critical Care Medicine, Lokmanya Hospitals,
Chinchwad, Pune, India Code Number: cm05016 Abstract We present a rare case of unexpected sudden death in a young woman with undiagnosed sickle disease. The provocative factors for the terminal events were excessive exercise in the form of trekking, urinary infection, and emotional stress .the sudden cardiovascular collapse could have resulted from acute pulmonary hypertension resulting from severe hemolysis or acute chest syndrome .Keywords: Anxiety, Hemolysis, Sickle Cell Disease, Sudden Death, Trekking, Urinary Infection Introduction Sudden deaths in recently married females are usually a cause of speculation. The cause of death could be both natural and unnatural. It is traumatizing for the husband and family members. The situation is aggravated if the patient is from outstation and the past history is not available. We are presenting this case of a young lady who had an unexpected sudden death. The non-specific nature of her complaints and the paucity of clinical signs misguided the clinicians on the potentially lethal outcome. The clue to her diagnosis lay in the peripheral smear examination. [Figure - 1]Case Report A 27 years old apparently healthy female, from Madhya Pradesh married for 7 days was on a honeymoon to a hill station in Maharashtra. She had trekked for 6-8 km daily during the last 5 days and complained of joint pains for which a local practitioner had given NSAIDS. Late that night she was admitted by him with pain in abdomen, breathlessness and severe anxiety. Her BP then was 100/70 mm of Hg, PR:100/mm, rest of the clinical examination was normal. Her lab investigation such as hemogram [Hb 10 gm/dl], blood urea and serum creatinine and x-ray chest, ECG was done and they were normal. She continued to be extremely restless. She was given Inj. diazepam, tramadol, dexamethasone, ceftrixone and was transferred here with a provisional diagnosis of Rheumatic Disease / anxiety. At 6.45 a.m, on arrival in our E. R. she was conscious, oriented and anxious. She complained of pain in abdomen and breathlessness. There was a past history of arthritis - details of which was not known. She was not on any regular treatment. On arrival her BP was 100/70 mm of Hg, PR 110/min, RR:32/min, she appeared markedly pale and icteric. Her cardiovascular examination was normal except for tachycardia. On auscultation of the chest there were minimal bilateral basal crepts. Abdomen was soft and spleen was just palpable. Saturation was 92% on room air and on oxygen 98%. She was given antibiotics, H2 blocker, nasal O2 and routine lab investigation was sent for. Her USG abdomen done at 8.45 am - revealed mild mucosal thickening of gall bladder, splenomegaly. Rest of the organs appeared normal. There was no fluid collection in the pleural, pericardial or peritoneal space. Her ECG was normal except for sinus tachycardia, X ray chest was normal. ABG could not be done. Blood grouping and cross matching was asked for and other lab investigations showed - Hb:5.3 gm/dl, WBC count:36,700 /cu mm with 76% neutrophils. Blood sugar 144 mg/dl, Urea 79 mg/dl, S. Creatinine 1.4 mg/dl, S.Na 134 mEq/lit., S.K 4.3 mEq/lit., S. Chloride 95 meq/lit., S. Bilirubin 5.0 mg/dl, conjugated 2.3 mg/dl and unconjugated 2.7 mg/dl, Alkaline phosphatase 297 IU/lit., SGPT 130 IU/lit., SGOT 248 IU/lit. Urine was turbid with trace albumin, 5 -10 pus cells and bacilli 3 +, few granular casts. Peripheral Smear examination showed neutrophilic leucocytosis, decreased platelets, microcytes+, anisopoikilocytosis, nucleated and few sickle shaped RBC′s and Spherocytes. Haemoglobin Electrophoresis was advised. At 10 a.m. patient had unexpected sudden witnessed cardiorespiratory arrest and could not be resuscitated. Autopsy was asked for. Hemolytic anemia study reports received later revealed - direct coombs test - Negative, Sickling (Na metabisulphite) - Positive, Solubility Test - Positive, Haemoglobin A2 factor - 0.90% (Normal range upto 3.80%), Foetal Hb - 23.10% (Normal range 1%), Haemoglobin S/D/G Factor - 74.20%, Haemoglobin A1 factor - 1.80% (Normal range 95-99%) which is suggestive of sickle cell anemia. The post-mortem report (personal communication) showed splenomegaly, icterus and no infarct in any organs, histopathology report was not available. Discussion Once sickle cell disease [SCD] was considered as a diagnosis, our patient′s symptoms of joint pains, dyspnoea, anxiety, pallor and jaundice could be explained. In India SCD is seen mainly in the tribal belts of central India.[1] Our patient belonged to the same area. From the meager history available, she had virtually asymptomatic childhood and was apparently never diagnosed as having SCD. Sonography did not reveal any gallstones and there was no evidence of joint swellings, deformities or leg ulcers suggesting chronic disease. SCD is remarkable for its clinical heterogeneity. Some patients have repeated episodes of admissions while others, will be totally asymptomatic.[2] Although everyone with SCD shares the same gene mutation, there are five recognized haplotypes based on its origin. They are Bantu, Arab-India, Senegal, Benin, Cameroon and Central African Republic.[2] It is a well-established fact that SCD has varying severity of clinical presentation in different population groups.The Central African patients have the worst prognosis while those from Senegal have the least severe form of the disease.[2] There is a dearth of data on SCD in India as compared to that in Africa and America. Leg ulcers and priapism are said to be uncommon, while splenomegaly is common in Indian SCD.[1] This is unlike most African or American patients who have non functional small spleens due to repeated infarcts.[1],[2] Besides the haplotypes, two other factors, which affect the severity of the disease are, the presence of fetal hemoglobin [HbF] and concurrent alpha thalassemia.[3] Platt et al[4] who examined predictive factors for life expectancy and risk factors for early death among black Americans felt HbF greater than 8.6 % improved survival. In Kar et al study, of patients from Orissa state in India, protective levels of HbF averaged 16.64 %. This higher average level of fetal hemoglobin probably explains the less severe disease in Indians and Arabs.[5] Steinberg et al observed that females tend to have higher levels of HbF than males irrespective of haplotypes.[3] As there is a heterogenecity in HbF distribution in the red cells, percentage of red cells having HbF is equally important in preventing the sickling of the red cells. Fetal hemoglobin levels of more than 25% in a pancellular distribution may be required to eliminate intracellular polymerization and hemolysis.[6],[7] However there are other studies that have found no correlation between HbF levels and severity of disease, suggesting that HbF in conjunction with other moderating factors determine the severity of the disease in patients of SCD. Seltzer et al observed five black families with high HbF levels who had varying degree of severity even in patients with similar HbF levels.[8] Our patient had a preterminal HbF level of 23.1 gms /dl which is consistent with Kar et al findings in Indian patients.[5] Whether this factor contributed to her previous asymptomatic status cannot be commented upon with certainty. Our patient had a very short history of illness lasting for few hours. She had evidence of acute hemolysis, abdominal pain, breathlessness and anxiety. This fits the description of sickle crisis. Vasoocclusive crisis can lead to life threatening major organ infarcts but usually is not a cause of sudden death. Our patient′s clinical examination, sonography and autopsy findings did not suggest any major organ infarcts. Therefore major organ infarct due to vasoocclusive crisis, alone cannot explain her sudden death within hours of admission. Acute chest syndrome [ACS] is a severe and catastrophic complication of SCD and is characterized by chest pain, tachypnoea, fever, cough and arterial oxygen desaturation. Young patients may have repeated episodes, leading to cor pulmonale. It can be severe with reported deaths of 1.8% in children and 4.3% in adults[9],[10] and is presumed to occur due to in situ sickling within the lung, producing pain and temporary pulmonary dysfunction. The most frequent findings are rales. Infiltrates may be seen in the chest x-ray even though quite often it may be normal.[10] The symptoms of ACS may be preceded by an acute decrease in hemoglobin concentration ,a decrease in platelet count and an increase in nucleated red cells.[10] The risk factors for development of ACS in patients with SCD are earlier age, a lower concentration of HbF, higher steady haemoglobin concentrations and higher steady state WBC counts.[10],[11] Our patient had a normal chest x-ray. Although there was no history of fever, chest pain or cough in our patient, she had rales and complained of breathlessness and had evidence of severe hemolysis. Under these circumstances a diagnosis of acute chest syndrome cannot be totally ruled out. Even if she had ACS it cannot totally explain sudden collapse. Moreover she did not respond to the oxygen or ventilatory support. In addition to this exchange transfusions have been advocated in the treatment of acute chest syndrome.[9] Incidentally WBC count can vary considerably in sickle cell crisis and need not always represent infection. WBCs and reticulocytes play a major role in pathogenesis of sickle cell crisis.[2] The urine was turbid with pus cells and bacteria indicating urinary infection. Her clinical picture however indicated sudden cardiovascular collapse. The persistent hyperdynamic circulation of critical anemia can affect heart in sickle cell anemia. A left sided heart disease has also been described as a complication of SCD.[12] Cardiac autonomic dysfunction has been reported to occur in 58% of SCD in one study, and may also be a risk factor for sudden death.[13] Pulmonary hypertension and chronic lung disease are the two most common causes of death among SCD patients. Retrospective studies have reported as many as 60% of the patients being affected by pulmonary hypertension.[14] Factors that contribute to lung injury include infection, bronchoreactive lung disease and fat embolism.[14] Mild undetected episodes of regional pulmonary hypoxia are reported to be important in development of pulmonary hypertension and sudden death syndrome.[6],[14] Routine clinical and laboratory findings are insensitive to detect pulmonary hypertension. Clinical symptoms and chest radiographs may be normal till irreversible damage has occurred although high-resolution computed tomographic scanning demonstrates micro vascular occlusion and hypoperfusion of the lungs.[12],[14] In their large prospective study of pulmonary hypertension in SCD patients, Gladwin et al, found echocardiography as a reliable noninvasive screening test for detection and identification of high-risk group for pulmonary hypertension.[6] A tricuspid regurgitant jet velocity of at least 2.5 m/sec was an indicator of pulmonary hypertension. They also concluded that in comparison with patients who have primary pulmonary hypertension, patients with SCD have a higher risk of death with mild elevation in pulmonary pressure. Exchange transfusion has been advocated in the treatment of acute chest syndrome.[15] The plasma hemoglobin present during hemolysis is reported to decrease the levels of both arginine and nitric oxide that have a cytoprotective and vasodilatative effect. This allows the process of development of pulmonary hypertension to go unchecked [14],[16] Clinical syndrome of hemolysis associated pulmonary hypertension is suggested by the fact that pulmonary hypertension develops in most forms of hemolytic anemia. This pulmonary hypertension may be a major cause of sudden deaths in patients with sickle cell disease in absence of coronary artery disease.[6] Most importantly a normal chest x-ray, normal ECG and normal pulse oxymetry reading can not rule out ACS or ominous development of PH in the presence of SCD.[14] Provocative factors for sickle cell crisis include infection, fever, excessive exercise, anxiety, abrupt changes in temperature and hypoxia. Except for fever all the above factors were seen in our patient. Exercise has been associated with sudden deaths, in patients with sickle traits.[17] Our patient had been trekking for five days at higher altitude than what she was used to. This was also associated with emotional stress of marriage, excessive traveling and urinary infection. We believe all these factors precipitated her sickle cell crisis and may have contributed to regional hypoxemias in the lung or even ACS. This along with the severe hemolysis could have resulted in acute pulmonary hypertension leading to sudden cardiovascular collapse from which patient could not be resuscitated. During her present illness she was given fluids, analgesics, steroids and oxygen therapy, all of which are recommended in treatment of sickle crisis but could not prevent the cascade of events which resulted in sudden unexpected death. Exchange transfusions, which have been reported to improve survival, unfortunately could not be done due to paucity of time. Conclusion Critical to good management is an approach that recognize most SCD patients reporting crisis symptoms do indeed have crisis or another significant medical problems. We are reporting this as a rare case of sudden death due to sickle cell death in a previously undiagnosed SCD in a young woman of Indian origin. Early recognition of this potentially fatal condition is crucial because intensive supportive therapy and early institution of exchange transfusion may reverse early pulmonary hypertension thereby preventing a catastrophic eventReferences
Copyright 2005 - Indian Journal of Critical Care Medicine The following images related to this document are available:Photo images[cm05016f1.jpg] |
| |||||||||

{kind=link}