 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
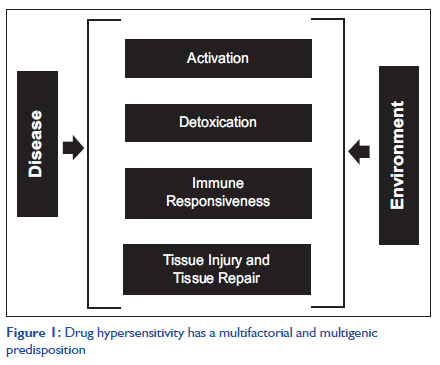
Indian Journal of Critical Care Medicine, Vol. 15, No. 3, July-September, 2011, pp. 173-175 Case Report Genetic predisposition to oxcarbazepine induced Stevens-Johnson syndrome Pranay Wal1, Ankita Wal2, Umeshwar Pandey3, Awani K Rai2, Anil Bhandari1 1 Jodhpur National University, Rajasthan, India DOI: 10.4103/0972-5229.84904 Abstract Stevens-Johnson syndrome (SJS) is a rare immunologic reaction that may involve skin or various mucosal surfaces. The etiology may range from multiple pharmacologic agents to viral infections. Associated findings can range from minimal skin and mucosal involvement to extensive dermal exfoliation, nephritis, lymphadenopathy, hepatitis, and multiple serologic abnormalities. We report a female patient of 38 years with a history of drug allergy who was administered oxcarbazepine for the management of right partial bronchial seizure due to left parasagittal mass lesion following which she developed papular rashes all over the body and diagnosed as SJS. Although carbamazepine (CBZ) is the most common cause of SJS, a new anticonvulsant, oxcarbazepine, which is structurally related to CBZ, has been shown to induce SJS.Keywords: Hypersensitivity, oxcarbazepine, Stevens-Johnson syndrome Introduction Stevens-Johnson syndrome (SJS) is thought to be a hypersensitivity complex affecting the skin and the mucous membranes. A new anticonvulsant, oxcarbazepine, which is structurally related to carbamazepine (CBZ), was introduced for use in patients with epilepsy. Drug hypersensitivity reactions can occur with most drugs, although the frequency, severity, and clinical manifestations vary. Drug hypersensitivity can be defined as an inappropriate immune response leading to tissue damage from an otherwise nontoxic agent. Drug hypersensitivity reactions to benzodiazepines often fall into the category of type B (or bizarre) adverse drug reactions, according to the classification proposed by Rawlins and Thompson. [1] The incidence of hypersensitivity varies according to the drug, the disease being treated, and the ethnicity of the patient. [2] Drug response (including drug hypersensitivity) is a multifactorial and multigenic process, dependent on a complex interaction between multiple genes and the environment [Figure - 1]. Each gene contributes to the risk of developing the hypersensitivity reaction, but each individual gene is neither necessary nor sufficient by itself to cause the reaction. [3] Case Report A female patient of 38 years with a history of drug allergy was complaining of fever, sore throat, and fatigue, at the time of admission. She was administered oxcarbazepine for the management of right partial bronchial seizure due to left parasagittal mass lesion following which she developed papular rashes all over the body and diagnosed as SJS. In this case, during the first week, we used 600 mg/day oxcarbazepine for seizure control, and then increased the dose after 3 days to 900 mg/day. Both the initial and titration doses were slightly lower than the recommended doses. After 10 days of treatment, ulcers and other lesions begin to appear in the mouth and lips along with the genital and anal regions. Ulcers in the mouth are usually extremely painful and reduce the patient′s ability to eat or drink. High fever and multiple maculopapular rashes were found over the patient′s face and neck initially on the 12 th day after taking oxcarbazepine. Two days later, some blisters were also observed on her thigh. Then, multiple oral ulcers and hyperemic conjunctivae were also noted. She was brought to our emergency department and admitted under the presumed diagnosis of SJS. Laboratory investigations showed leukocytosis (WBC, 14660/μL; reference value, 4,000-10,000/μL), and elevated C-reactive protein (59.30 μg/mL; reference range, 0-5 μg/mL). After obtaining informed consent, we carried out genotyping and took photos of the patient. HLA genotyping showed HLA-BFNx011518/BFNx014001. A skin biopsy was also performed. The stratum corneum appeared to be normal. There was marked liquefactive degeneration in the lower half of the epidermis with some dyskeratotic keratinocytes. The dermis showed predominant CD8+ lymphohistiocytic infiltration around the blood vessels and scanty eosinophils. The skin pathology finding was consistent with SJS. After corticosteroids (IV dexamethasone 1.0 mg/kg body weight) and antihistamine treatment for 10 days, the patient improved and was discharged from the hospital. Discussion Anticonvulsant hypersensitivity syndrome is a potentially fatal drug reaction with cutaneous and systemic reactions (incidence, 1 in 1000 to 1 in 10,000 exposures) to the arene oxide-producing anticonvulsants-Phenytoin, CBZ, and Phenobarbital sodium. CBZ and its derivatives are widely used anticonvulsants that can cause rashes in up to 10% of patients, and in occasional cases, this may be the precursor to the development of a hypersensitivity syndrome characterized by systemic manifestations such as fever and eosinophilia. [4] The diagnosis of SJS is based on clinical manifestations with acute onset of rapidly expanding targetoid erythematous macules, necrosis and detachment of the epidermis along with erythema, erosions, and crusting of two or more mucosal surfaces. [5] Our patient had skin targetoid erythematous rashes and mucosa involvement 2 weeks after starting oxcarbazepine treatment. During these 2 weeks, she took no medicine except for oxcarbazepine. The skin pathology finding revealed lymphohistiocytic infiltration around the blood vessels and scanty eosinophils, which was consistent with SJS. The patients usually develop a hypersensitivity reaction between 2 and 12 weeks after starting medicine. [6] It has been postulated that metabolites and not the parent drug are the causal agents. One of the first reports showed that HLA-BFNx011502 was present in 100% of CBZ-induced patients with SJS but in only 3% of patients tolerating CBZ and in 9% of the general population. [7] Other studies have confirmed these results in Han Chinese and in the Thai population. [11] In this case, we used a dose of 600 mg/day and then titrated it to 900 mg/day. It has been reported that higher daily doses of drugs are associated with an increased risk of SJS than lower doses, which is the case for allopurinol.[8] There is considerable debate about whether to treat SJS with systemic steroids. However, Lam et al. found that the early use of short-term systemic steroids for 3-5 days lacked any significant side effects and did not increase mortality or morbidity. [9] Our patient was treated with intravenous dexamethasone and antihistaminics for 10 days. She improved and was discharged. No sequels were found during 3 months of follow-up. Compared with other categories of drugs, such as antibiotics and NSAIDs, antiepileptic therapies are associated with a high incidence of SJS and toxic epidermal necrolysis (TEN). [10] Case reports by Chen et al. suggest that identification of such genetic factors [11] is important, not only to realize the prospect of developing preventive strategies but also to learn about the mechanisms of these reactions, which may ultimately lead to other preventive strategies through better drug design and to better treatment strategies for patients who develop the reactions. References
Copyright 2011 - Indian Journal of Critical Care Medicine The following images related to this document are available:Photo images[cm11043f1.jpg] |
| |||||||||

{kind=link}