 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Cancer, Vol. 40, No. 4, (October -December 2003) , pp. 144-147 Case Report Chronic Myeloid Leukemia Presenting with Absence of Basophils and Marked Dyspoiesis Anand M, Kumar R, Kumar L,* Barge S,* Singh S** From the Unit of Laboratory Oncology and *Department of Medical Oncology, Institute
Rotary Cancer Hospital, All India Institute of Medical Sciences, New Delhi, India;
and **Department of Pathology
Lady Hardinge Medical College, New Delhi, India.
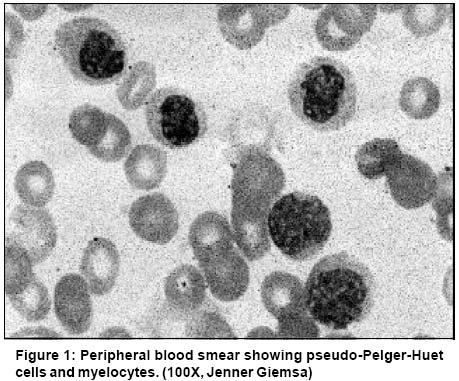
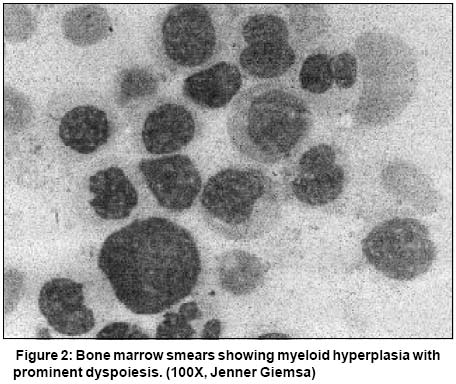
Code Number: cn03025 ABSTRACT: A 61-year old woman presented to us with fever, weakness and ecchymotic patches for one year. She had leucocytosis, anemia and thrombocytopenia. Peripheral blood smear showed 62% neutrophils, 32% myelocytes and metamyelocytes, 2% promyelocytes, 1% blasts, 2% monocytes, 1% lymphocytes but no basophils and marked dyspoiesis. Bone marrow picture was essentially the same. A diagnosis of atypical chronic myeloid leukemia was suggested. The correct diagnosis of chronic myeloid leukemia - accelerated phase was, however, made on cytogenetic analysis which showed Philadelphia chromosome (Ph) and isochromosome 17q [i(17q)]. This case describes a rare and diagnostically difficult presentation of CML arising out of a combination of prominent dyspoiesis and near absence of peripheral blood basophils. Key Words: CML, aCML, Basophil, Dyspoiesis. Introduction Laboratory diagnosis of chronic myeloid leukemia (CML) is generally a straightforward matter. Presence of myelocyte bulge combined with characteristic basophilia, allow the diagnosis to be easily made in most cases on peripheral blood smear (PBS) examination.1,2 We report a case where the diagnosis of CML was initially missed by us on peripheral blood morphology, because of the rare presentation with severe dyspoiesis and more importantly, paucity of basophils. Case Report A 61-year-old female patient presented with fever, weakness and bluish patches for 1 year. Physical examination revealed pallor, splenomegaly 4 cm and ecchymotic patches on the legs. Her hemoglobin was 70 g/l, TLC 50 x 109/l and platelets 26 x 109/l. Peripheral blood smear examination showed 62% neutrophils, 1% lymphocytes, 17% myelocytes, 15% metamyelocytes, 2% promyelocytes, 1% blasts, 2% monocytes and no basophils (<1/500 WBC). In addition, there was marked dysmyelopoiesis seen in approximately 35% of granulocytic series of cells, characterized by pseudo-Pelger-Huet anomaly (hyposegmented neutrophils with abnormally increased chromatin condensation) and a few hybrid (myelomonocytic) cells (Figure 1). Bone marrow smears showed a cellular preparation with marked myeloid hyperplasia (M:E ratio 99:1), prominent dyspoietic changes and blasts less than 2%. Megakaryocytes were reduced and basophils constituted <1/500 of the marrow cells (Figure 2). A diagnosis of atypical CML (aCML) was made on morphology. Leucocyte alkaline phosphatase (LAP) and cytogenetic studies were carried out for confirmation of diagnosis. LAP score was zero. Cytogenetic studies were performed on bone marrow aspirate by standard techniques using GTG banding. Ten metaphases were examined and showed a karyotype of 46XX, Philadelphia (Ph) chromosome in 30% of metaphases along with isochromosome 17q [i(17q)]. Demonstration of BCR-ABL fusion gene by RT-PCR/FISH was, however, not carried out. In the light of cytogenetic evidence, i.e., presence of Ph chromosome as well as i(17q), thromobocytopenia unrelated to therapy and presence of dyspoiesis, the diagnosis was revised to CML accelerated phase (CML-AP). The patient was offered Gleevec but declined to take it owing to the high cost involved. He was put on hydroxyurea but returned a day later with severe pain in both lower limbs. The TLC at this time was 20 x 109/L. She suffered a massive gastrointestinal bleed and died a few hours later. Discussion
The salient peripheral blood findings in this patient at the time she presented to us were leucocytosis (50 x 109/l), anemia, thrombocytopenia, mature and immature granulocytes showing marked dysmyelopoietic changes, 2% monocytes, essentially no basophils (<1/500 WBC) and <5% blasts. Bone marrow changes were qualitatively similiar. In addition there was mild dyserythropoiesis but no dysmegakaryopoiesis. The findings were therefore of both chronic myeloproliferation and myelodysplasia. In the earlier, French American British (FAB), recommendation,3 myeloproliferation and myelodysplasia defined separate disease groups, and patients with features of both, were placed in one of these; for example, myelodysplastic syndrome in the case of chronic myelomonocytic leukemia (CMML). The recently introduced WHO classification is less restrictive in this regard and has created a separate disease category called myelodysplastic/myeloproliferative diseases1,2 for conditions such as atypical CML (aCML), CMML and juvenile chronic myelomonocytic leukemia, that have features of both myeloproliferation and myelodysplasia. The WHO classification also recognizes that myeloproliferation and myelodysplasia may come to coexist when a well-recognized myeloproliferative disease such as CML, develops dysplasia and ineffective hematopoiesis in the course of its natural progression.2 Hence, based on the peripheral blood and bone marrow findings, the diagnoses we considered, were aCML, CMML and CML-accelerated phase (AP). Two features of our patient that call for discussion, while considering CML as a possible diagnosis, were prominent dyspoiesis and low number of circulating basophils. Dyspoietic features are unusual in CML chronic phase (CP) and if present, suggest an incorrect diagnosis or progression of disease.1,2,4-7 Though used in clinical practice, the value of dyspoiesis as an indicator of disease acceleration is, however, limited compared to certain other multivariate analysis-derived criteria,8,9 such as peripheral blood blasts ( >15%), blasts + promyelocytes >30%, basophils >20%, thromobocytopenia <100x109/L unrelated to therapy and cytogenetic clonal evolution. More importantly, from our perspective, prominent dyspoiesis does not pose a significant problem in recognizing CML. This is because the real nature of the disorder is revealed by the increased basophils and very low percentage of monocytes. Thus in our patient we did not consider CML accelerated phase as a possibility because of the near absence of basophils (<1/500 WBC). Basophils are an integral part of CML1,2,5and are rarely absent in chronic phase.4,10 In the widely quoted study of Spiers et al,10 hematological profile of 50 patients of Ph positive CML were studied and absolute basophilia was a consistent finding in all cases while morphological abnormality of the neutrophils was uncommon. In another study, hematological profile of 50 Ph positive CML patients were studied and basophilia was documented in all but one patient.9 In a sequential multiparameteric study of individuals exposed to atomic bomb radiation in Japan, it was shown that increase in basophils is observed after the appearance of Philadelphia chromosome and precedes such features as thrombocytosis, decreased LAP, appearance of immature granulocytes, etc.11 Basophils can be regarded as the sine qua non of the diagnosis of CML and in their absence, such a diagnosis can be validated only if supported by cytogenetic and molecular evidence.1,5,12 Our second diagnostic consideration was CMML. A defining feature of CMML is an absolute monocyte count of >1 x 109/l, almost always due to >10% monocytes. Neutrophil precursors usually account for <10% of the WBC.2,6,13 Though our patient had an absolute monocytosis of >1 x 109/l this was more on account of the high TLC (50 x 109/l) and not due to the percentage of monocytes, which was low (2%). Also, neutrophil precursors (35%) were prominent in the peripheral blood. We therefore ruled out CMML on morphology. Based on peripheral blood findings of leucocytosis due to increased number of neutrophils, prominent dysgranulopoiesis, neutrophil precursors >10%, basophils <2%, minimal absolute monocytosis with <10% monocytes in the DLC, together with corroborative findings in the bone marrow2 we made a diagnosis of aCML. Low LAP score did not argue against this as low LAP has been reported in most cases of acml.1 In view of this we did not regard a score of zero as being decisively in favour of CML. Cytogentic analysis being a mandatory part of the work-up of cases such as ours, was done subsequently and showed quite unexpectedly, Ph chromosome, confirming a diagnosis of CML. Isochromosome 17q was also present. This is the third most common secondary karyotypic abnormality seen in CML in transformation (clonal evolution) including 9-29% cases of CML in blast crisis14,15 and is also one of the commoner chromosomal anomalies seen in aCML.1,14,16 In the present case since the karyotyping was carried out one year after the onset of symptoms, we cannot be certain whether this additional cytogenetic abnormality was present right from the beginning or acquired later in the course of disease. Its presence in our patient who had practically no basophils was of particular interest as this cytogenetic change has been reported to be associated with marked basophilia.14 Conclusion We conclude that a peripheral blood and bone marrow picture of severe myelodysplasia together with myeloproliferation may exceptionally be due to CML even in the absence of the all-important basophil. Diagnosis in such cases cannot be based on morphological features alone and mandate cytogenetic and/or molecular studies. References
Copyright 2003 - Indian Journal of Cancer The following images related to this document are available:Photo images[cn03025f1.jpg] [cn03025f2.jpg] |
| |||||||||

{kind=link}
{kind=link}