 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
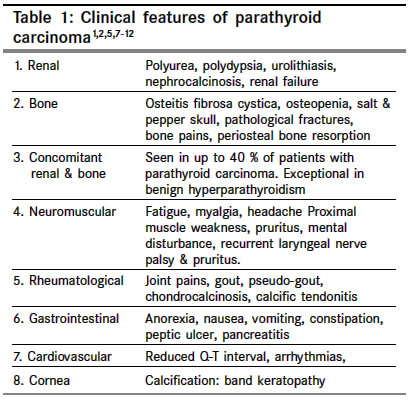
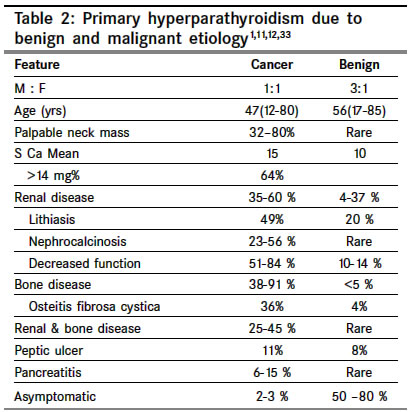
Indian Journal of Cancer, Vol. 41, No. 2, Apr-Jun, 2004, pp. 51-59 Review Article The carcinoma of parathyroid gland Kulkarni PS, Parikh PurvishM Department of Medical Oncology, Tata Memorial Hospital, Parel, Mumbai Code Number: cn04010 ABSTRACT Parathyroid carcinoma constitutes less than 1% of primary hyperparathyroidism. The exact etiology is not known. Prior radiation to neck, chronic renal failure and genetic factors are thought to play a role. The male to female ratio is one. Parathyroid carcinomas are slow growing, have a tendency to recur locally and metastasize late. 95% of parathyroid carcinomas are functioning. The major distinguishing features of malignant hyperparathyroidism are presence of a palpable mass in the neck and features of severe hypercalcemia. By far the most important test to diagnose primary hyperparathyroidism is serum level of Immunoreactive PTH. The diagnosis of primary hyperparathyroidism is essentially clinical and biochemical. Biopsy is not necessary before definitive surgery. CT scan appears to be the best investigation for detecting the primary tumor, its local extent and metastases. Most of the symptoms are attributable to hypercalcemia, which needs to be treated aggressively. Early surgery with 'en bloc' resection of the tumor is the only potentially curative treatment. Parathyroid carcinoma is traditionally said to be resistant to radiotherapy. Various chemotherapeutic agents have been used with partial anecdotal responses. The 5-year survival is about 50% and 10-year survival varies from 13-49%. Keywords: Primary hyperparathyroidism, parathyroid carcinoma, management Parathyroid tumors account for a very small percentage of all head and neck neoplasms. Among the cases of primary hyperparathyroidism about 85% of the patients have a single adenoma, 5% have multiple adenomas, 10% have hyperplasia, and less than 1% have carcinoma.[1] Parathyroid carcinoma is therefore a rare cause of parathyroid disease, whose exact incidence is unclear. The first case of parathyroid carcinoma described by de Quervain (1904) was a non-functioning carcinoma. Sainton and Millet were the first to describe functioning carcinoma of the parathyroid gland in 1933.[2] The incidence of hyperparathyroidism has increased since 1974. Because most of these tumors retain the ability to produce active parathyroid hormone, hypercalcemia is a common feature. After the introduction of multi-channel autoanalyzer biochemical features of hyperparathyroidism are increasingly detected in asymptomatic individuals.[3] Heath et al have shown that the average annual incidence of primary hyperparathyroidism has increased from 8/1,00,000 to 28/1,00,000 following the introduction of routine serum calcium measurements among hospital admissions. In comparison to the benign causes of hyperparathyroidism, the number of parathyroid carcinoma is essential unchanged. However, there has been a small increase in the proportion of patients presenting with mildly symptomatic parathyroid cancer following the detection of hypercalcemia on biochemical screening.[2],[3],[4] Various series in west show that the incidence of parathyroid carcinoma among patients with primary hyperparathyroidism is probably around 1%. This was also confirmed from Mainland China where the incidence was also1%. However, the nationwide survey of parathyroid disease in Japan (Ishida et al) has documented an incidence of nearly 5%.[4],[5] The cause for this is unclear and may represent either an absolute (due to genetic or environmental factors) or a relative increase (due to large numbers of patients with mildly symptomatic primary hyperparathyroidism). In a recent Italian study, 5.2% of patients operated for primary hyperparathyroidism were found to have parathyroid carcinoma.[3],[4],[5] Approximately, 400 patients of parathyroid carcinoma have been reported, till now, in the English literature.[6],[7],[8],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18],[19],[20],[21],[22],[23] ETIOLOGY The etiology of parathyroid carcinoma is unknown. Previous radiation to neck is a known risk factor for head and neck cancer. It has been reported in 9% to 30% of patients with hyperparathyroidism and benign parathyroid tumors. Average latency period is 37 years. However only about five parathyroid cancer patients with a history of previous cervical radiation have been reported. In them, radiation doses of 30 to 60 Gy were delivered 5-35 years before the development of the parathyroid cancer.[1],[11],[20],[22] Chronic renal failure, which leads to secondary hyperparathyroidism, has also been implicated in few patients with parathyroid carcinoma. One patient had coexisting parathyroid carcinoma, adenoma and hyperplasia while another had a history of both chronic renal failure and prior radiation to neck. Berland et al have reported the first case of parathyroid carcinoma developing in a patient on hemodialysis. Since then about 13 cases in patients on hemodialysis have been reported in English literature. The average age of these patients was 49 years. Only 50% of these presented with signs of hypercalcemia. No preoperative features are known to distinguish hemodialysis patients with parathyroid carcinoma from those with parathyroid hyperplasia. Interestingly, their clinical course may be less aggressive due to effect of renal insufficiency on lowering serum calcium levels.[1],[11],[23] Despite earlier conflicting results, it now appears that parathyroid adenomas /hyperplasia in MEN type I are monoclonal neoplasms. There have been case reports of carcinoma occurring within an adenoma / hyperplasic parathyroid gland or in association with celiac disease. However, the relevance of these findings is unclear.[1],[6] Parathyroid carcinoma has been reported in families with Autosomal dominant familial hyperparathyroidism indicating an increased risk associated with this disorder.[1] Increased risk of parathyroid carcinoma is associated with hereditary hyperparathyroidism-jaw tumor syndrome (HPT-JT). Germ-line inactivating HRPT2 mutations have been found in HPT-JT and there is evidence that they may be of pathogenic importance in sporadic parathyroid carcinoma also.[24],[25],[26] In addition, 3% of cases of parathyroid carcinoma have been reported in patients aged less than 20 years- indicating that genetic factors may play an important role in this cancer.[1],[2],[11],[27] MOLECULAR PATHOGENESIS Cyclin D1 or PRAD1 (parathyroid adenoma1) is an oncogene located at chromosome 11 band 11q13. A chromosomal rearrangement of the cyclin D1 gene involving the regulatory region of the PTH gene has been reported in 5% of parathyroid adenomas. In addition, the cyclin D1 oncoprotein is overexpressed in 18-40% of parathyroid adenoma.[2],[11],[27] Overexpression of cyclin D1 protein is strikingly frequent in parathyroid carcinomas. Cryns et al showed that all (11/11) parathyroid cancers studied lacked an RB allele and most had complete absence of nuclear staining for the RB protein.[28] Subramanian et al, using a mouse monoclonal antibody, were able to show evidence of RB gene inactivation in 2 of 3 cancers and only 1 of 11 adenomas. Pearce et al found allelic deletions of the 13q12-14 region involving both the RB gene and the BRCA2 gene in 3 of 19 patients′adenomas with aggressive clinical and histopathological features and 1 parathyroid carcinoma.[11],[28],[29] Similar losses involving 13q have also been reported by other investigators. These data strongly support the presence of a tumor suppressor gene on the long arm chromosome 13, which is critical to the parathyroid carcinogenesis. Tumor specific gains or losses of chromosomal material suggest that oncogenes (at location 1q, 5q, 9q, 16p, 19p,and Xq) and tumor suppressor genes (at locations including 1p, 3q, 4q, 13q,and 21q) may be involved in the pathogenesis of parathyroid carcinoma.[11],[27],[29],[30],[31],[32] Interestingly, a number of genes commonly lost in adenomas were rarely lost in carcinomas. This supports the hypothesis that parathyroid carcinomas tend to arise de novo rather than from pre-existing adenomas.[11] CLINICAL FEATURES The male to female ratio in most series is 1:1[in primary hyperparathyroidism due to benign causes there is a marked female preponderance (ratio of 3-4: 1)].[1],[11],[33] The mean age of the patients with this disease is 45 years with a range from 12 to 80 years [primary hyperparathyroidism due to benign causes occurs almost a decade later].[1],[4],[11] However, considerations of gender and age are of little help in evaluating the etiology and diagnosis in individual patient. Parathyroid carcinomas are typically slow growing tumors. They have a tendency to recur locally and metastasis occurs late in its natural history. 95% of parathyroid carcinomas are functioning i.e. secrete parathyroid hormone.[8],[11] Hence, almost all the patients are symptomatic at presentation. The clinical features of parathyroid carcinoma are primarily due to the effects of excessive secretion of PTH by the functioning tumor (rather than to infiltration of vital organs by tumor mass) [Table - 1]. This is of immense help to the clinician in differentiating parathyroid carcinoma from its much more common benign counterpart. Clinical features of primary hyperparathyroidism due to benign causes differ markedly from those caused by the cancer as shown in the following [Table - 2]. The major distinguishing features of malignant hyperparathyroidism are presence of a palpable mass in the neck and features of severe hypercalcemia.[1],[8],[11] Hoarseness with recurrent laryngeal nerve involvement is rare. However its presence in a patient with primary hyperparathyroidism, who has not had prior neck surgery, is very suggestive of parathyroid carcinoma.[1] Parathyroid carcinoma has been reported during pregnancy. Hypercalcemia can have serious consequences on the fetus. Both maternal and fetal morbidity and mortality is significantly increased. Surgical treatment prior to delivery during 2nd or 3rd trimester is therefore recommended.[1],[34] LABORATORY FEATURES Hypercalcemia, the hallmark of hyperparathyroidism, is usually severe in patients with parathyroid carcinoma. The serum calcium level >14 mg/dl is seen in about two thirds of the patients (as compared to less than 10% patients with benign hyperparathyroidism). Anemia is also more common in carcinoma (up to 80% versus 10% in benign). Other laboratory features include hypophosphatemia, hypercalciuria,
and hyperphosphaturia.[1],[7],[11],[12] Increased S. Cl and decreased bicarbonate can lead to metabolic acidosis. This will aggravate hypercalcemia by reducing the binding of calcium to albumin and increasing the dissolution of the bone mineral. Urinary cyclic AMP is elevated due to PTH binding to renal receptors. In case of metabolic bone disease (without metastases), S alkaline phosphates as well as urinary excretion of hydroxyproline is increased.[1],[11],[12] By far the most important test to diagnose primary hyperparathyroidism is serum level of Immunoreactive PTH. Different radioimmunoasays are available directed either at the intact molecule, the active fragments (amino- or N-terminal) or at inactive fragments (mid region, carboxyl- or C-terminal region). Elevated levels of S. imunoreactive PTH (iPTH) are virtually diagnostic and can be very high (3-10 times above the upper limit of normal for the assay employed) in cases of functioning parathyroid carcinoma.[8],[11] HISTOPATHOLOGY The normal parathyroid gland contains chief cells, oxyphill cells, water-clear cells and adipose tissue.[1] The carcinoma arising from the oxyphill cells is extremely rare and has been reported in case report form.[35] The majority of the parathyroid carcinoma arises from the chief cells. It is often difficult to differentiate between benign or malignant nature of these tumors. [7],[35] Parathyroid carcinoma appears as a grayish, hard lobulated tumor (compared to soft, round to oval, reddish brown appearance of the adenoma). It is surrounded by dense fibrotic capsule. Its adherence to and invasion of surrounding structures is an important sign of malignancy. This is seen in about 40-50% of cases. The average size of the carcinoma is 3 cm; with a mean weight of 6 to 12 gm. Microscopically the cells resemble ′watermelon′seeds and are larger and better defined than adenomatous cells. The cells are bland and uniform with cellular atypia being rare.[7],[11],[12] Castleman and Shantz described the criteria for diagnosing the malignancy as presence of 1) trabecular pattern with thick fibrous bands, 2) nuclear palisading, 3) mitotic figures, 4) capsular and blood vessel invasion. However, these features are neither conclusive nor constant.[2] McKeown et al have challenged these criteria and showed that cellular atypia and pleomorphism are not reliable in endocrine malignancies and mitotic activity in parenchymal cells must be distinguished from that involving endothelial cells. Similarly, care must be taken in assessing invasion as nests of parathyroid tissue in the capsule may represent benign entrapment. Locally recurrent lesions must also be interpreted with caution, as rupture of benign adenomas is known to seed the operative field with viable cells, which can lead to recurrent disease. Because of these issues, a number of investigators have recommended documented metastatic disease as the only definite evidence of malignancy.[36] Hence Bondeson et al carefully reviewed the histological specimens from carcinomas proven on the basis of documented metastatic disease. They found that the mitotic activity in carcinoma was variable- with 50% having rates similar to those seen in benign lesions. Fibrosis was present in 80% of carcinomas, usually with a fibrous capsule and thick hyaline bands throughout the parenchyma. However, 20% of cancers did not show this fibrous band pattern. Also, trabecular architecture was not indicative of carcinomas being seen in parathyroid adenoma and hyperplasia. Up to 70% of carcinomas had nuclear atypia and up to 11% carcinomas consisted of oxyphil cells instead of chief cell morphology. The best predictors of malignancy were presence of invasion, fibrotic capsule, and nuclear atypia.[37] Thus, an important conclusion is that when a carcinoma is suspected on clinical ground and/ or gross appearance, the surgeon should resect the tumor en bloc without relying on biopsy or frozen section Several other histological techniques have been investigated to further improve the accuracy of diagnosing parathyroid carcinoma. Electron microscopy of malignant cells reveals nuclear and mitochondrial alterations and evidence of increased secretory activity, but does not help to distinguish between benign and malignant lesions.[1],[7],[35] Electron microscopic study of the oxyphill cell carcinoma is characterized by cells packed with mitochondria.[35] Mean nuclear DNA content is greater and an aneuploid DNA pattern is more common in parathyroid carcinoma than in adenomas and when present aneuploidy is associated with a poorer prognosis.[7] However aneuploidy occurs too frequently in parathyroid adenoma to be of any help to differentiate it from carcinoma. Immunohistochemical staining of RB protein may be of some help to differentiate in parathyroid carcinoma and adenoma. Cryns et al first reported that staining for RB protein with polyclonal antibodies was commonly absent in parathyroid carcinomas and was almost always present in adenomas.[28] After this initial report, various other investigators have reported results supporting these results. However there have been conflicting reports as well. Abbona et al found significant differences in Ki-67 staining between benign and malignant parathyroid disease.[38] Hence the value of Immunohistochemical studies for differential diagnosis of parathyroid lesions is still not settled. Invasive growth of various neoplasms may be facilitated by tumoral secretion of proteolytic enzymes. Farnebo et al reported that gelatinase-A messenger ribonucleic acid (m-RNA) was detected in 14 of 18 unequivocal and 4 of 13 equivocal parathyroid cancer. The strongest signal, as expected, was detected in the fibroblasts and macrophages at the tumor border, rather than in the tumor cells. This relatively new technique may provide additional support for the diagnosis of malignancy.[1],[7] IMAGING In benign hyperparathyroidism, first time exploration by an experienced surgeon will successfully detect the tumors in more than 90% of the cases. Imaging plays a more important role in recurrent or residual disease after initial surgery. They are also useful before initial surgery when carcinoma is suspected on clinical grounds [Table - 3]. It allows evaluation of the local extent of the tumor as well as the metastases to the cervical nodes-thereby helping the surgeon to plan the extent of surgery.[39],[40] Non-invasive Imaging Esophagogram with careful evaluation of the cervical esophagus can indirectly evaluate the parathyroid glands. USG with its high resolution and real time technique is an excellent investigation. However, its accuracy is highly operator dependant and it cannot completely assess the retroesophageal, retrotracheal and mediastinal areas. Computed tomography, Radionuclide scintigraphy (particularly sequential use of Thallium-201, which concentrates both in parathyroid and thyroid, and Technetium 99m, which concentrates in the thyroid followed by subtraction allows imaging of the parathyroid tumors) and MRI can be used. Their comparative sensitivity and specificity is as follows:[41],[42],[43],[44],[45] Thus, for patients suspected to have parathyroid carcinoma, CT scan appears to be the best investigation for detecting the primary tumor, its local extent and metastases. None of the imaging modalities has a sensitivity of more than 50% for small tumors.[7],[38] The use of flow cytometric analysis to determine aneuploidy, S-phase fraction, and proliferation index values to distinguish parathyroid carcinoma from adenoma or hyperplasia has yielded conflicting results. Production of human chorionic gonadotropin (hCG) subunits alpha and beta has been observed in parathyroid carcinoma and not in benign tumors.[7],[39] Invasive Imaging
Venography with venous sampling for iPTH from thyroid, vertebral, thymic and internal mammary veins remains one of the most sensitive techniques. A unilateral gradient is in favour of a single adenoma or carcinoma, whereas a bilateral gradient usually indicates diffuse hyperplasia.[39] It is a time consuming but relatively safe procedure. Selective or superselective angiography requires experienced personnel. Instances of severe neurological complications (e.g. quadriplegia, and even death) can occur due to inadvertent injection of contrast material into spinal branches of the thyrocervical or costocervical trunk. Safer but less sensitive techniques include non-selective intraarterial or intravenous digital subtraction angiography.[39],[40] Staging It is interesting to know that American Joint Committee on Cancer/ International Union Against Cancer (AJCC/UICC) has not yet developed a TNM staging system for parathyroid carcinoma because these are fairly rare tumors. Shaha and Shah have proposed a staging system, which might help in uniform reporting of the extent of disease in patients with parathyroid carcinoma.[46] T1
= Primary tumor < 3 cm Stage Grouping
Stage I: T1N0M0 TREATMENT Surgery It is the only potentially curative treatment for parathyroid carcinoma.[8] Early surgery (when extensive local invasion and distal metastases are less likely) is the most important factor for optimal outcome.[39] For this reasons both preoperative suspicion and intraoperative recognition of the malignant nature of the tumor are of paramount importance. Patients whose clinical presentation is suggestive of parathyroid carcinoma warrant thorough exploration of all the four parathyroid glands, as parathyroid carcinoma has been reported to coexist with parathyroid adenoma or hyperplasia.[9] When the gross pathological findings suggest malignancy, En bloc resection of the tumor is mandatory.[1],[8],[9] The ipsilateral thyroid lobe and isthmus along with all areas of local adherence should be removed. The trachea should be skeletonized and the tumor should be removed without violating the capsule. If recurrent laryngeal nerve is involved, it should be sacrificed as failure to do so may lead to local recurrence. Simple biopsy with /without frozen section should not be attempted (due to inability to distinguish between benign and malignant lesions as well as fear of causing seeding of the surrounding tissues).[8] Often, the surgeon might have to rely on the gross appearance of the lesion to determine its nature. The En bloc resection is associated with a 10% local failure rate with 90% of the patients being long-term survivors. On the contrary, incomplete resection leads to a local recurrence rate of 50% and disease related mortality of 46%.[1],[7],[39] A difficult situation may arise when the diagnosis of the malignancy is done in the early postoperative period on the basis of pathology. It is particularly complex given the notorious uncertainty surrounding the histology of parathyroid carcinoma. If the gross finding were suggestive of a parathyroid cancer and the subsequent pathology appears to be aggressive or the patient remains hypercalcemic, re-exploration of the neck is indicated. If none of these features is present, but the diagnosis is made on the basis of the microscopic characteristics, immediate re-operation may not always be necessary. Such a patient must be observed carefully with frequent measurements of PTH and serum calcium levels.[1],[7],[11],[12],[39] There is no role of prophylactic neck dissection.[39] When distinction between benign and malignant nature of the lesion is in doubt, the other ipsilateral parathyroid should be removed to ascertain the possibility of diffuse hyperplasia.[1],[7] Close post-op monitoring is essential to detect hungry bone syndrome. It should be regarded as a sign of a successful surgery and is caused by deposition of calcium and phosphate into the bones. Its hypocalcaemia may be severe and protracted, requiring large doss of intravenous calcium and calcitriol- which needs to be anticipated.[39] Parathyroid carcinoma grows slowly and metastasizes late. Early recurrence is an unfavorable prognostic factor.[7],[12] Most recurrences usually occur within 2 to 3 years of initial surgery. Local recurrence (30%) as well as metastases to cervical lymph nodes (30 to 40%) are fairly common. Distal metastases, when occur, commonly involve the lungs (20 to 40%).[1],[8],[12],[39] Other sites like mediastinum, bone, pleura, pericardium and pancreas also may be affected. The disease is usually indolent and most patients die of metabolic complications rather than disease. Even a very small metastatic deposit may produce significant PTH to cause severe hypercalcemia. Hence significant palliation may be achieved from the resection of the recurrent or residual disease.[8] In patients with recurrent hypercalcemia, localization studies should be performed before re-operation. Thallium 201- technetium 99m scanning is useful in locating tumors in the neck and the mediastinum.[41],[42] Intra-operative localization of the parathyroid may be achieved with the help of technetium 99m-sestamibi used concurrently with a handheld, ã-detecting probe.[39],[42],[43] CT scan and MRI may be useful adjuncts to ultrasonography in evaluation of the disease in the neck and are better for detecting distant metastases in the chest or abdomen. If non-invasive testing does not localize the tumor, selective venous sampling or arteriography may help.[40] Systemic therapy Radiotherapy Parathyroid carcinoma is traditionally said to be resistant to radiotherapy (isolated reports of long-term control do exist).[10],[39] The dose of radiation recommended is 40 -50 Gy in patients with high risk of recurrence i.e. patients with microscopic/ macroscopic residual disease after surgical resection. The radiation field should include tumor bed and extend superiorly to the hyoid bone and inferiorly to the clavicle. The field should also include paratracheal and perithyroidal areas. For patients with lymph node involvement, the radiation target volume should include all cervical lymph nodes, including the superior mediastinum. Radiation may also find a place in the palliative management of the patients with metastatic disease.[10],[22],[47],[48] Lillemore and Dudley reported in 1985 that by delivering postoperative adjuvant radiation to 3 patients, with a follow-up of 16 to 49 months, none of the patients had evidence of recurrence. Chow et al (1998) reported 6 patients with adjuvant RT. All the patients tolerated radiation well with minimal toxicity. After a mean follow up of 62.3 months, none of the patients had any evidence of recurrence.[61] In contrast, Shortell et al (1991) reported recurrence in all 3 patients treated with parathyroidectomy alone.[22],[39] Chemotherapy Due to the rarity of the parathyroid carcinoma there are no large randomized chemotherapy clinical trials. Hence the experience of use of various chemotherapeutic agents is limited- usually to anecdotal cases.[39] Several regimens (nitrogen mustards; vincristine, cyclophosphamide, and actinomycin D; adriamycin, cyclophosphamide, and 5-flurouracil; and adriamycin alone) have been tried and found to be ineffective.[1],[8],[10],[39] Partial and temporary remissions have been reported with estrogen and testosterone. A PR of 10 months has been reported with ′Hexestrol′-a synthetic estrogen. Cisplatin (single agent 50 mg/m2 was used in a recurrent disease without response.[39] A single patient with pulmonary metastases responded to treatment with dacarbazine, 5-flurouracil and cyclophosphamide with a decrease in PTH and normalization of S. Ca++ for 13 months.[2] Another patient responded to dacarbazine alone with a brief but significant decline in serum calcium level. The ′MACC′(methotrexate, adriamycin, cyclophosphamide and CCNU) regimen produced a dramatic regression of large mediastinal mass and malignant pleural effusion lasting 18 months.[39] Another case of non-functioning carcinoma was treated with a modified MACC (mitoxantrone used instead of adriamycin) protocol and had a partial response for 101 months.[7],[39] Over last few decades, many new drugs with novel mechanisms of action and better toxicity profile have become available. However, there have been no studies evaluating the efficacy of these agents in parathyroid carcinoma. Thus, the role of chemotherapy in this disease entity is not well defined and needs further investigations.[7],[10],[39] Treatment of hypercalcemia When parathyroid carcinoma has become widely disseminated and surgical resection is no longer effective, the prognosis is poor.[1] The therapeutic goal at this point is to control the hypercalcemia- a Herculean task. The hypercalcemia due to parathyroid carcinoma is to be treated in the same way as hypercalcemia due to any other cause. Management includes infusion of saline, loop diuretics and various hypocalcemic agents. These include bisphosphonates, plicamycin, calcitonin and gallium nitrate. Recently a novel calcimimetic agent (NPS R568) has been developed as an allosteric modulator of the calcium receptor. It was used successfully to control hypercalcemia in a patient with parathyroid carcinoma. The S. Calcium was controlled for 2 years without any adverse effects.[49],[50] Other agents like WR-2721 (Amifostine) and octreotide have been used in some cases.[1],[7] A novel approach to the therapy of parathyroid carcinoma induced resistant hypercalcemia was immunization with human and bovine PTH peptides. Production of antibodies against self-peptides requires T-cell help; however, thymic deletion of self-reactive T cells normally precludes the development of T-cell help after immunization with the self-antigens. In this case, the T-cells recognize foreign antigens (bovine PTH) and help B cells to produce antibodies against self-PTH (Molecular Mimicry). Antibodies against PTH were detected in 4 weeks time and there was rapid improvement in the symptoms without significant adverse effects.[51]Non-functioning Parathyroid Carcinoma This rare entity perhaps represents 1.9-5% of parathyroid carcinomas.[7],[8],[11] Murphy et al have reported a total of 12 such cases in the English literature. The median age of the patients was 50 years (range 27 to 71 years). These patients have no hypercalcemia and S iPTH is within normal limits.[1],[39] The factors responsible for the lack of hyperparathyroidism may be lack of hormone synthesis, impaired or reduced secretion of the hormone or synthesis of an abnormal hormone. Impaired/reduced secretion of the hormone seems to be the most likely cause. Electron microscopy shows lipid, glycogen and neurosecretory granules in cytoplasm, thus distinguishing them from thyroid or renal cell carcinoma. These tumors need to be distinguished from other non-functioning parathyroid carcinomas and metastatic carcinomas. These tumors are treated identical to ones with functioning carcinomas and carry a similar prognosis (median survival 2 years with a range from 9 months to 5 years).[1],[7],[8]Prognosis Prognosis of patients with parathyroid carcinoma is variable. Early recognition and complete resection at the time of initial surgery carries the best prognosis. The average time between the initial surgery and the first recurrence is approximately 3 years with a recurrence free survival of 30%.[1],[7],[11],[39] Once the tumor has recurred, complete cure is unlikely. However prolonged and meaningful survival may be possible with palliative surgery and/or chemotherapy. In such cases, the 5-year overall survival is about 50% and 10-year survival varies from 13 to 49%.[1],[7],[39]REFERENCES
Copyright 2004 - Indian Journal of Cancer The following images related to this document are available:Photo images[cn04010t2.jpg] [cn04010t1.jpg] [cn04010t3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}