 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
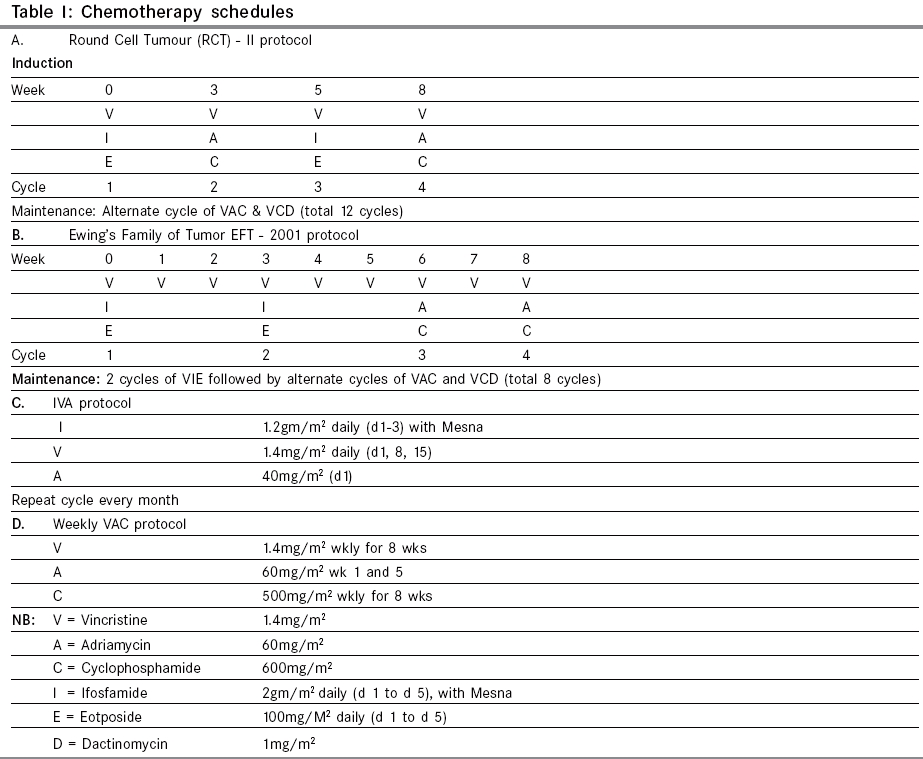
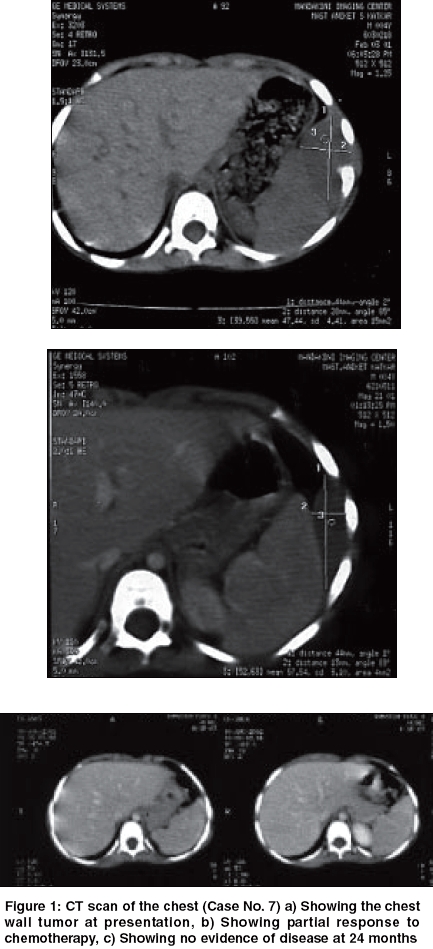
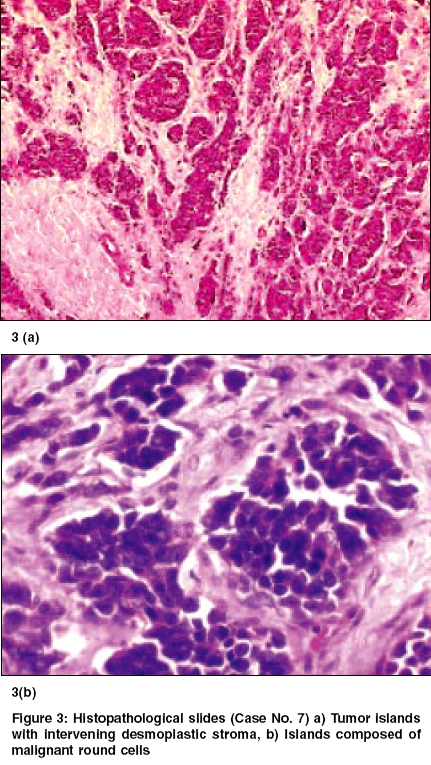
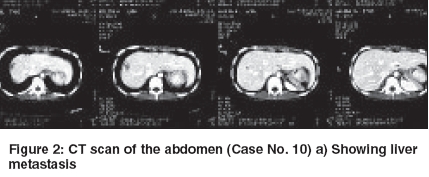
Indian Journal of Cancer, Vol. 42, No. 2, April-June, 2005, pp. 78-84 Original Article Desmoplastic small round cell tumor: Extra abdominal and abdominal presentations and the results of treatment Biswas G, Laskar S, Banavali SD, Gujral S, Kurkure PA, Muckaden M, Parikh PM, Nair ChandrikaN Department of Medical Oncology, Tata Memorial Center, Mumbai Code Number: cn05015 Abstract BACKGROUND: Desmoplastic small round cell tumor (DSRCT) is a rare malignant neoplasm of adolescent males. Current multimodality treatment prolongs life and rarely achieves cure. Keywords: Desmoplastic round cell tumor, Management and outcome Introduction Desmoplastic small round cell tumor is a rare malignant neoplasm. It is characterized by a distinct immunohistochemical profile and a recurrent, specific, chromosomal translocation. It is an aggressive and often misdiagnosed neoplasm of children and young adult. [1],[2] This tumor has predilection for involvement of the peritoneum. CT features are frequently multiple bulky heterogeneous and necrotic soft tissue masses in the abdomen, usually without any obvious organ base. It is sometimes associated with ascites, adenopathies and liver metastases. Typically, tumors consist of small, undifferentiated cells or spindle cells invested within an abundant desmoplastic stroma. Immunohistochemical studies show polyphenotypic differentiation with expression of epithelial, neural and muscle markers. [1] A recurrent specific chromosomal abnormality t (11:22) (p13:q12) has been reported in DSRCT.[3] The breakpoints in this translocation involve two genes EWS, which is altered in the t (11:22) (q24:q12) rearrangement characteristic of the Ewing′s family of tumors and WT1, which is a Wilms tumor gene. EWS-WT1 chimeric transcripts are considered diagnostic of this disease. [4],[5] Current treatment protocols include multiagent chemotherapy and adjuvant surgery and radiotherapy. [6],[7] Several chemotherapy regimens including alkylating agents and aggressive chemotherapy regimen followed by myeloblative therapy and stem cells rescue have been tried.[8] However, the emphasis is on achieving a complete and durable response. We report our experience in the management of DSRCT at our center. Materials and Methods 18 patients diagnosed as DSRCT at our institute for management during the period 1994 to 2005 are included this analysis. The diagnosis was based on routine histopathology and the radiological findings. Histopathological evaluation of tumor was done using H & E Microscopy and Immunohistochemistry (IHC). IHC was done for eleven cases. Complete physical and radiological evaluation and metastatic workup using CT scan Thorax/Abdomen/Pelvis, Bone scan, Bone marrow aspiration and biopsy was done for most patients. 6 patients had metastatic disease at presentation. The chemotherapy schedules received by the patients are documented in [Table - 1]. At the end of 8-9 weeks of induction chemotherapy, patients were evaluated for local treatment when feasible. Surgery alone or in combination with radiotherapy was used for local treatment. This was followed by maintenance chemotherapy. The outcome and survival of these patients are documented in [Table - 2] (C). Results The clinical, radiological, treatment received and the outcome and survival of these patients are shown in the [Table - 2] (A, B, C). The age ranged from 1½ year to 30 years. Ten patients were in the pediatric age group ( < 15 years) and seven were females. Fifteen patients presented with abdomino-pelvic swellings, of which 3 had ovarian mass. One patient had chest wall swelling (Case no. 7) and another patient had an ulcerative mass in the palm of the right hand (Case no. 9). There was no bone marrow involvement among 15 patients done. Bone scan was abnormal in one patient among the 11 patients done. LDH was raised in 6 out of 8 cases done, which probably signify disease load. In cases 13 and 15 we did pre-treatment CA.125, which were significantly raised. We could perform diagnostic EWS-WT1 in case no.17 was positive. The response seen with chemotherapy was 39% (CR-1, PR-6). One had Stable disease and rest was considered to have Progressive disease including those not evaluable for response. In total there were 8 surgeries (Outside-4, TMH-4). There were only 3 complete excision including negative cut margins. 4 patients received radiotherapy including one that received Stereotactic technique. The details of the three unusual primary site of involvement (Case no: 7, 9 & 18) are as follows: Case no. 7 Case no. 9 Case no. 18 Response was seen in 7 (n = 18) patients. CR achieved in one and PR in six patients. One had stable disease. Seven had documented progressive disease and 3 patients were not evaluable for response and have been considered to have progressive disease. Complete excision of the mass was also possible in case no 3. It was found to be an ovarian mass and there was no residual viable tumor. She was continued on 2 more cycles of chemotherapy. At 48 months, she was found to be disease free. Case no. 10 underwent upfront surgery. She had excision of the ovarian mass, excision of mesentery along with the nodules. The liver metastasis also showed response to chemotherapy. The liver metastasis [Figure - 2] a was treated with using Extracranial Stereotactic Radiotherapy technique delivering a dose of 28Gy in 4 # @ 7Gy per # x 2 #/week. At 41 months (March 2005) she is alive and disease free. Case no. 14 presented with ascitis, peritoneal deposits, liver and splenic involvement. Post EFT-2001 protocol induction, he attained PR. Subsequently, completed EFT-2001 protocol maintenance. Post planned treatment PET scan showed diffuse uptake in liver and one focus in spleen. He was then started on oral chemotherapy with Etoposide, Thioguanine and Tamoxifen. Overall survival has been poor except in the 3 cases where complete excision was possible. Other cases had disease progression locally. The median overall survival is 6.5 months (Range 1-57 m). DISCUSSION We are reporting unusual sites - chest wall, hand, thigh and ovary besides the common sites and the clinical outcome. Similar to all the series reported so far, the abdomino-pelvic site was the commonest site of presentation. Of these 3 cases primarily seems to be arising from the ovary. Other sites Pleura, Para-testicular, Lung, Ovary, thorax, intracranial, soft tissue, bone of the hand and Sino-nasal regions have been reported.[9],[10],[11],[12],[13],[14],[20] DSRCT is commonly reported in children and young adults and male predominance is noted. [6] In our series also male predominance was seen. A high serum CA 125 level may be a specific marker for DSRCT, and thus may permit early diagnosis and treatment of this fast-growing tumor. [21],[22] We have done in 2 patients at baseline and it was found to be significantly raised. Most patients presented with extensive local disease. Distant metastases were seen in six patients. The most common site of distant metastases was Liver[18] (n=3/6). None had bone marrow involvement. The diagnosis of DSRCT may be suspected in young men with multiple bulky heterogeneous peritoneal soft tissue mass. Imaging is useful for staging and also for guided biopsies. [18] Markers for multilineage differentiation has been reported by Lae ME et al. [15] This group has found these tumors to be positive for Desmin (Dot pattern) in 81%, WT1 in 91%, Keratin in 87%, and Neuron Specific Enolase in 84% of the cases. The EWS-WT1 gene fusion transcript was detected in 29 of 30 times. Our patient (Case no. 9) showed polyphenotypic differentiation markers but did not show desmoplasia. However, the metastatic axillary mass later showed extensive desmoplasia along with polyphenotypic expression. They require aggressive multimodality therapy.[18] Intensive chemotherapy and complete excision of the tumor would be required to achieve long term disease free survival. Often surgery is not feasible especially in the abdomino- pelvic tumors due to the extensive involvement. Whole abdomino-pelvic irradiation (WAPI) has been reported by Goodman KA et al, [16] as a novel approach for the residual disease following aggressive chemotherapy and debulking surgery. However, the overall survival and relapse free survival rate reported by them at 3 years were 48% and 19% respectively. Aggressive multimodality treatment followed by high dose Busulphan with autologous rescue resulting in a disease free survival of only 19 months post transplant is also reported. [8] Successful clinical response was reported by Philip M Rosoft et al with Irinotecan and hence suggests trials with Irinotecan alone or in combination. [17] Current treatment prolongs life and rarely achieves cure. Neoadjuvant chemotherapy, greater than 90% tumor debulking, and radiotherapy have been shown to prolong survival. Future efforts must focus on cell-specific treatment protocols. [19] The demerits of this analysis are absence of specific cytogenetics, non-uniformity of protocol and few aggressive surgeries. Conclusion Though abdomino-pelvic site is the commonest site of DSRCT, this malignancy can occur at other sites which are not associated with serosal surface and thus the histogenesis of this tumor still remains obscure. Our experience shows that some degree of chemo-sensitivity is observed in DSRCT. However a complete surgical excision seems to improve survival. Results of treatment using current combined modality treatment remain unsatisfactory. There is a definite need to further risk stratify thus develop techniques and regimens for improving outcome. Acknowledgments We are thankful to Dr. S. V Kane, Professor, Pathology for doing the electron microscopy in case no 3.References
Copyright 2005 - Indian Journal of Cancer The following images related to this document are available:Photo images[cn05015t2.jpg] [cn05015f3.jpg] [cn05015f2.jpg] [cn05015f1.jpg] [cn05015t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}