 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
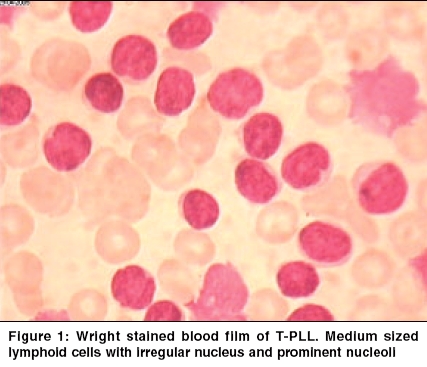
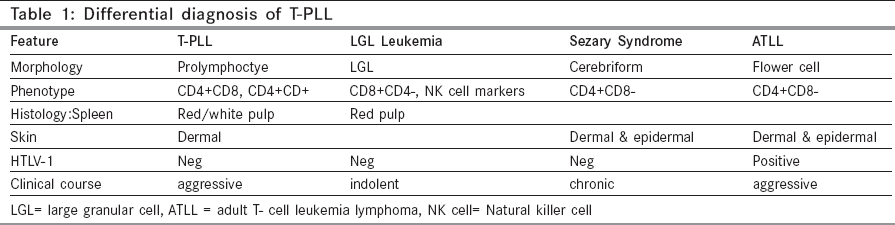
Indian Journal of Cancer, Vol. 42, No. 2, April-June, 2005, pp. 104-106 Case Reports T- cell prolymphocytic leukemia - A rare case Ghosh Sharmila, Advani SureshH Department of Hematologist, Asian Institute of Oncology, SL Raheja Hospital, Raheja Hospital Marg, Mahim, Mumbai Code Number: cn05021 Abstract T- cell Prolymhocytic leukemia (T-PLL) is a rare mature post-thymic T-cell malignancy that is usually reported in the elderly and follows an aggressive course. A 68 year old male presented with a history of weakness and weight loss of two months duration. Clinical examination revealed pallor, enlarged cervical and axillary lymph nodes and splenomegaly. He also had a maculo- papular skin rash. There was marked leucocytosis, anemia and thrombocytopenia (WBC 445 x103/ml, Hb 8.5gm/dl, Platelet 25x103/µl) with 60% prolymphocytes in the peripheral blood. Bone marrow was hypercellular with an excess of prolymphocytes. Flow cytometric analysis of the bone marrow showed positivity for CD2, CD3, CD4, CD5 and CD7. T- PLL is a rare T cell disorder with characteristic clinical and laboratory features.Currently, no optimal treatment exists although there has been some success with 2'- deoxycoformycin or Campath-1H Keywords: Prolymphocytes, Leukemia, T cell, Lymphoproliferative Introduction T-cell prolymphocytic leukemia (T-PLL) is a rare post-thymic T-cell lymphoproliferative disorder with distinct clinical, immunophenotypic and cytogenetic features. It accounts for one third of all post thymic T- cell leukemias. T and B PLL are two distinct diseases with different clinical and laboratory features. It is generally resistant to conventional chemotherapy and the median survival is 7.5 months. [1], [2] A precise diagnosis of this condition is important in determining optimum patient management and treatment. Here, we present a rare case of T-PLL in an elderly male who was treated with chlorambucil but was subsequently lost to follow-up. Case Report A 68 year old male presented with a history of weakness and weight loss of two months duration. Clinical examination revealed pallor, enlarged cervical and axillary lymph nodes and splenomegaly. He also had a maculo-papular skin rash. There was marked leucocytosis, anemia and thrombocytopenia (WBC 445 x103/ml, Hb 8.5gm/dl, Platelet 25 x 103/µl). The peripheral blood comprised of a uniform population of cells with 82% prolymphocytes in the peripheral blood. These cells were moderately sized lymphocytes with a high nuclear cytoplasmic ratio, moderately condensed chromatin and a prominent nucleolus. [Figure - 1] The cytoplasm was basophilic, scanty and agranular with occasional protrusions. Many of the cells had an irregular and folded nucleus. The bone marrow aspirate smears were hypercellular with an excess of prolymphocytes (76%). Flow cytometric analysis on the bone marrow showed positivity for CD2, CD3, CD4, CD5 and CD7. All the B cell markers were negative. Based on the morphologic features and the immunophenotypic profile, a diagnosis of prolymphocytic leukemia of T-cell phenotype was made. The patient was started on chlorambucil but was subsequently lost to follow up.Discussion T-cell PLL is a rare hematologic malignancy that was first described in 1973.[3] It accounts for approximately 3% of the T-lymphocyte disorders.[4] B-PLL and T-PLL are distinct disease entities with different clinical and laboratory features.[5] The disease affects adults with a median age of 65 years.[5] A slight male preference is noted. Patients usually present with splenomegaly, lymphadenopathy, hepatomegaly, skin lesions and marked lymphocytosis. Pleural effusion, ascites and central nervous system involvement may develop during the course of the disease.[1],[5] The white blood cell count is markedly raised usually over 100x 103/µl and one third of cases have anemia and thrombocytopenia. The predominant cell population in the peripheral blood comprises of lymphoid cells having features of prolymphocytes. Morphologically, the T prolymphoctes are smaller than the B prolymphocytes and have more cytoplasmic basophilia. The nucleus is irregular in half the cases of T-PLL which may be seen as short indentations or rarely as polylobated nuclei.[5],[6] Serum antibodies to the human T-cell leukemia virus-I and II have not been detected.[6] In 20% of T-PLL cases, the cells are smaller with more condensed chromatin. This is the small cell variant of T-PLL and they behave similar to the typical cases.[6] T-PLL must be distinguished from B-PLL as well as the other post-thymic T-cell malignancies. Clinical features such as lymphadenopathy and skin lesions are found in T-PLL. These along with immunophenotyping can differentiate T from B-PLL. The differential diagnosis from other T-cell malignancies is based on laboratory features such as morphology, histology and immunophenotyping. [Table - 1][5] T-PLL cells have a mature post thymic T-cell phenotype and do not express CD1a and terminal deoxynucleotidyl transferase (TdT). The cells are positive for CD2, CD5, CD3 and CD7 (strong). Occasional cases may be CD3/ cyto CD3 negative but the T-cell Receptor-beta/gamma chain genes are always rearranged.[5] A CD4+/CD8- phenotype is seen in approximately two-thirds of the cases, CD4+/CD8+ in about 25% cases and CD4-/CD8+ expression is less often seen. [1],[5] The intensity of CD52 expression, (seen in normal and neoplastic lymphoid cells as well as monocytes and macrophages), is stronger in T-prolymphocytes than normal T cells. [5],[6] This may explain the good response of T-PLL to anti CD52 therapy. The recurrent chromosomal abnormality most often seen in T-PLL is inv (14)(q11;q32).[7] The inversion (14) results in juxtaposition of a putative oncogene, TCL-1, located at 14q32.1, a region centromeric to the immunoglobulin heavy chain locus, with the gene coding for the TCR-alpha chain at 14q11 resulting in the expression and activation of TCL-1. Trisomy 8 and iso 8q are also common in T-PLL and is associated with c-myc overexpression. High expression of c-myc may play a role in disease progression as a secondary event. [5] T-PLL is an aggressive disorder with a median survival of seven months. Currently there is no approved standard treatment for T-PLL. Treatment with alkylating agents such as chlorambucil has been disappointing as patients are either resistant or achieve only a partial or short lived response. Although about one third of patients respond to CHOP chemotherapy (cyclophosphamide, doxorubicin, vincristine, prednisone), eventually the disease recurs in all of them. The purine analogue, 2′- deoxycoformycin (DCF) has been more successful in the treatment of this condition. Matutes et al have reported partial or complete response in 45% of their patients treated with this drug as a single agent. [5] Recently Campath-1H, an anti- CD52 humanised monoclonal antibody, has been found to be highly effective in T-PLL.[1],[5],[6],[8] Responses have been noted in approximately two thirds of patients including those who have been refractory to treatment with deoxycoformycin. An effective treatment schedule recommended by Matutes is as follows: Deoxycoformycin followed by Campath-1H in patients who achieve partial response or are refractory to DCF. Alternately Campath-1H may be used as the first line therapy.[5] These strategies allow the harvesting of peripheral blood stem cells and the use of high dose chemotherapy with an autograft in young T-PLL patients with the possibility of cure. References
Copyright 2005 - Indian Journal of Cancer The following images related to this document are available:Photo images[cn05021f1.jpg] [cn05021t1.jpg] |
| |||||||||

{kind=link}
{kind=link}