 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
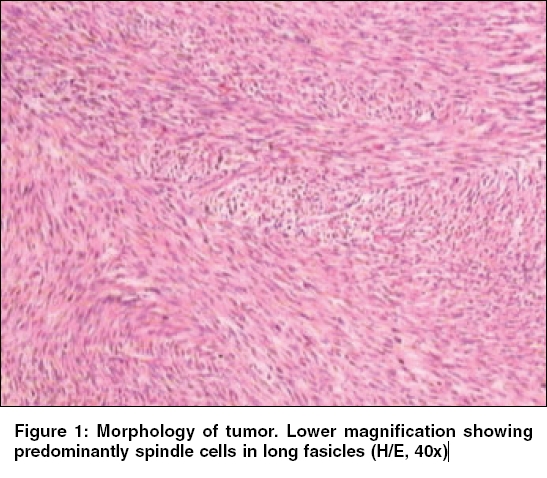
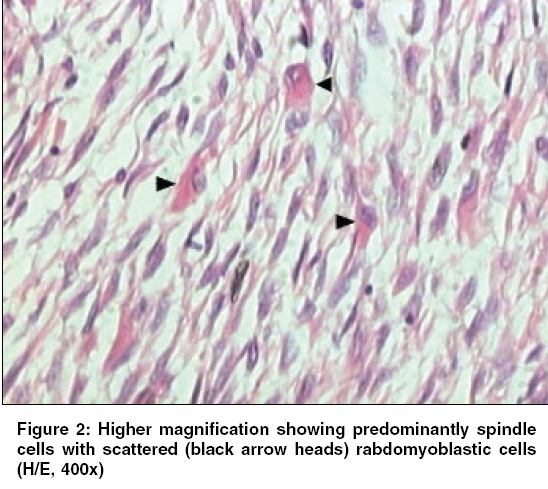
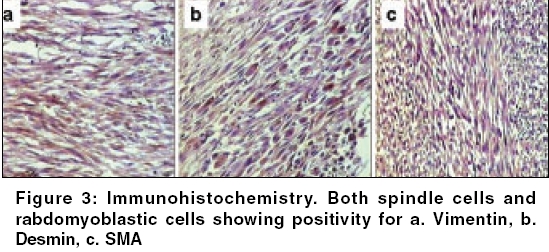
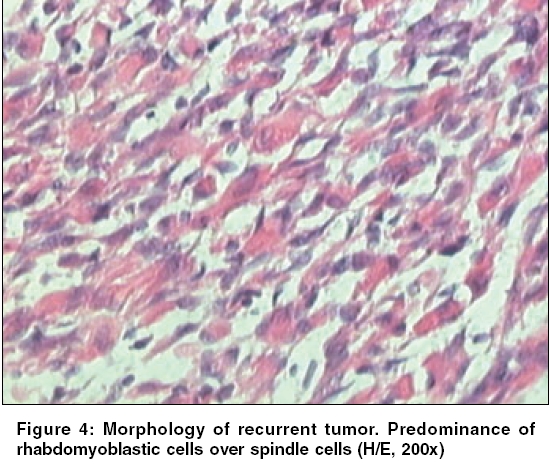
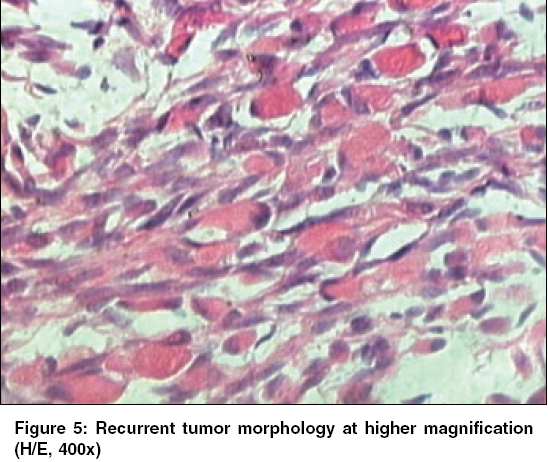
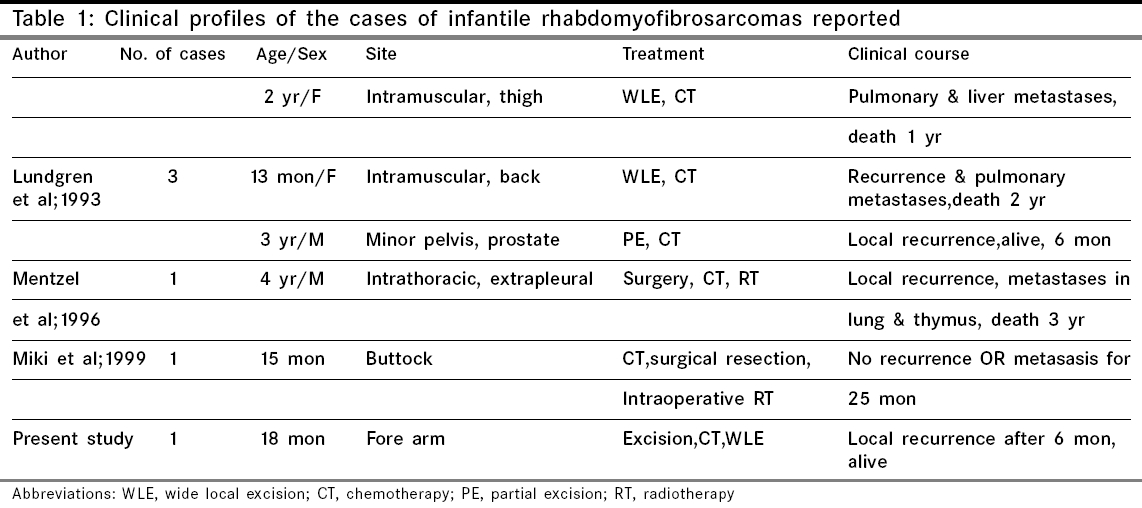
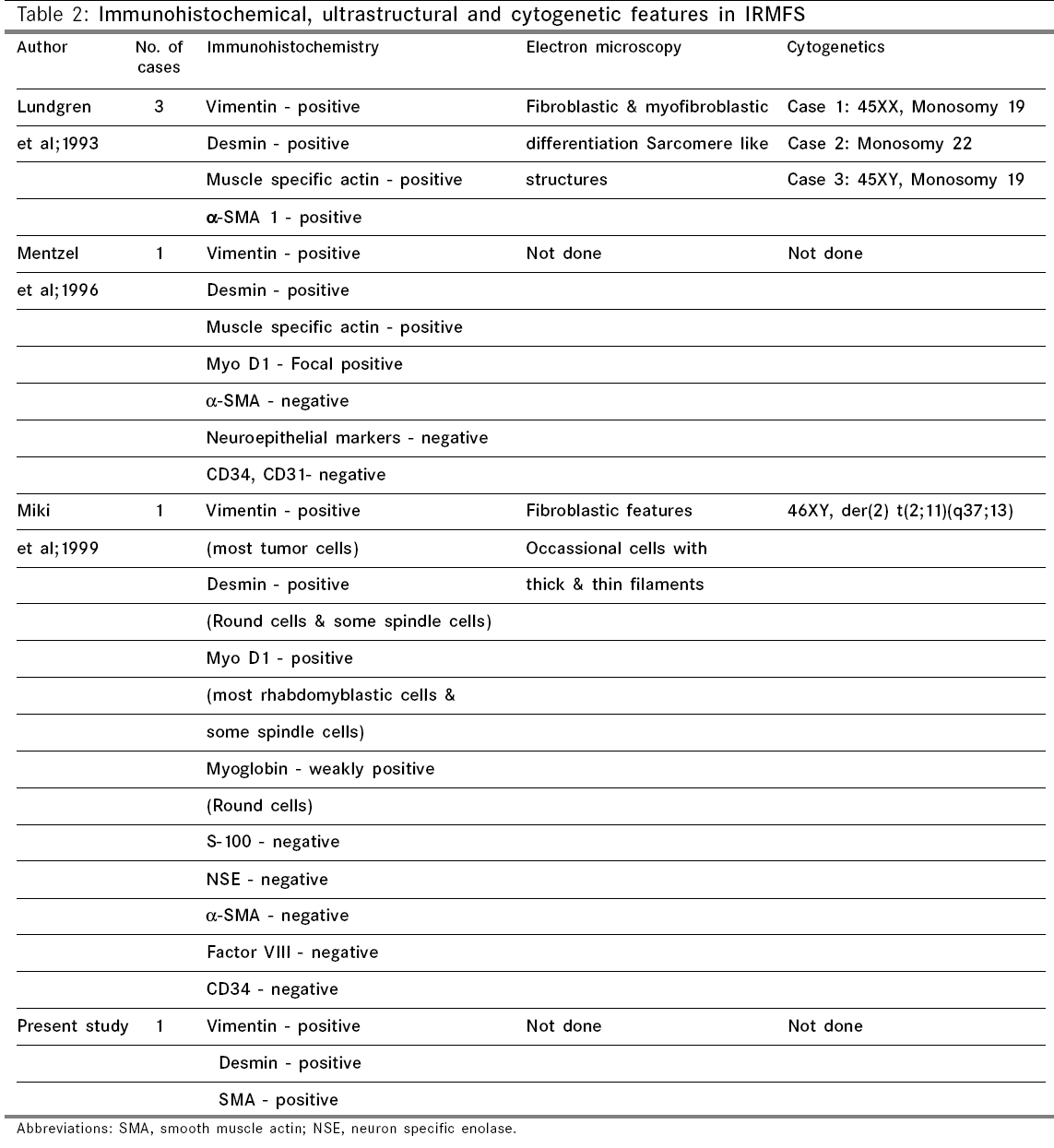
Indian Journal of Cancer, Vol. 43, No. 1, January-March, 2006, pp. 39-42 Case Report Infantile rhabdomyofibrosarcoma: A distinct variant or a missing link between fibrosarcoma and rhabdomyosarcoma? Rao Satish I, Uppin Shantveer G, Ratnakar KS, Sundaram C, Senthil Rajappa P* Departments of Pathology, *Medical Oncology, Nizam’s Institute of Medical Sciences, Hyderabad, India Code Number: cn06008 Abstract Infantile rhabdomyofibrosarcoma (IRMFS) is a rare soft tissue tumour affecting infants and young children. It occupies an intermediate position between infantile fibrosarcoma and spindle cell rhabdomyosarcoma in its clinical presentation, behaviour, morphology, immunohistochemical and ultrastructural features. This case is reported here to reiterate its occurence as tumour with distinct morphological immunohistochemical and clinical behavioral patterns.Keywords: Infantile rhabdomyofibrosarcoma, infantile fibrosarcoma, spindle cell rhabdomysarcoma. Introduction Childhood fibrous tumours continue to be enigmas especially those which do not have any adult counterpart. Accurate prediction of biological behaviour based on morphological features may be difficult leading to inappropriate therapy. We present such a case of infantile rhabdomyofibrosarcoma (IRMFS), the reports of which are scant in the literature.Case Report Clinical summary The patient received chemotherapy with six cycles of vincristine, cyclophosphamide and adriamycin at three weekly intervals. Post-chemotherapy MRI scan showed a recurrent tumor which was completely excised with wide resection margins. The sections from the same showed similar morphology as the original tumor with relative predominance of rhabdomyoblastic cells over the spindle cells [Figure - 4][Figure - 5]. There was no associated necrosis, pleomorphism or any mitoses in these sections also. The resected margins were grossly and microscopically free of tumor. Undifferentiated round to oval cells resembling the immature rhabdomyoblasts were not seen in either of two specimens. The patient is on follow up without any evidence of any further recurrence or metastasis since last two years. Discussion Infantile fibrosarcoma is a relatively rare tumour initially described by Stout in 1962. Which presents as a non-tender, pain less swelling with variable rate of growth.[1] This tumour mainly involves the extremities and head neck region with rare occurrence in retroperitoneum, mesentery and orbit.[2],[3],[4],[5] Microscopically it is composed of spindle cells arranged in long fascicles with minimal pleomorphism and mitoses. Spindle cell rhabdomyosarcoma occurs more commonly in paratesticular region and rarely affects head and neck.[6] Histologically it is composed of an exclusive population of elongated fusiform cells with cigar shaped nuclei and prominent nucleoli. Cytoplasmic cross striations may be seen. Lundgren et al (1993) identified for the first time IRMFS in three children. These were initially diagnosed as infantile fibrosarcoma, but two of the patients developed metastasis and died within two years of primary operation whereas the third patient was alive with local recurrence. This behaviour was contrary to the favourable course of infantile fibrosarcoma. An extensive comparitive immunohistochemical, ultrastructural and cytogenetic studies were done on these cases along with other cases of infantile fibrosarcoma.[7] Two more cases of IRMFS were subsequently reported.[8],[9] [Table - 1][Table - 2] highlight the various observations on all the cases of infantile rhabomyofibosarcoma reported in the literature. The present case is similar in many aspects that the patient is below 2 years with involvement of extremity and showing biphasic population of spindle cells admixed with polygonal rhabdomyoblastic cells. Both of these cells showed positivity for vimentin, desmin and focally for SMA in concordance with the previous studies. There was distinct absence of any undifferentiated small round to oval cells, any pleomorphism, necrosis or mitosis. The tumour recurred within six months of first surgery however the patient did not develop any metastasis. One notable feature in our case is that there was an overt increase of rhabdomyoblastic cells over the spindle cell population in recurrent tumour after chemotherapy. This feature has been observed in one case of IRMFS and some of childhood rhabdomyosarcomas following chemotherapy.[9],[10] Current molecular genetic studies report a characteristic translocation of t(12;15) (p13;q25) and the fusion transcript ETV6-NTRK3 detected by Reverse transcriptase polymerase chain reaction as diagnostic feature of infantile fibrosarcoma but specific cytogenetic abnormalities have not been reported in cases of spindle cell rhabdomyosarcoma.[11] In the above mentioned cases of IRMFS, monosomies of chromosomes 19 and 22 and der(2) t(2;11)(q37;13) were observed in three cases.[7],[9] These cytogenetic variations need to be supported studies on larger number of cases. The confirmatory categorization of the present case as IRMFS is limited by lack of cytogenetic and molecular studies. Conclusion Infantile rhabomyofibosarcoma occupies an intermediate position between an indolent behaving infantile fibrosarcoma and an aggressively behaving rhabdomyosarcoma. The number of cases of IRMFS reported in the literature are too small to arrive at a definite cytogenetic conclusion. The inadequacy of cases also hinders the correct categorization of this tumour. Hence these kind of cases need more diligent work up and close follow up.References
Copyright 2006 - Indian Journal of Cancer The following images related to this document are available:Photo images[cn06008f4.jpg] [cn06008t1.jpg] [cn06008f5.jpg] [cn06008f3.jpg] [cn06008f1.jpg] [cn06008t2.jpg] [cn06008f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}