 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
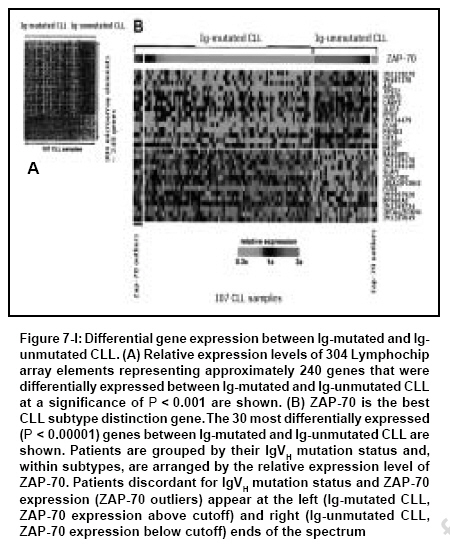
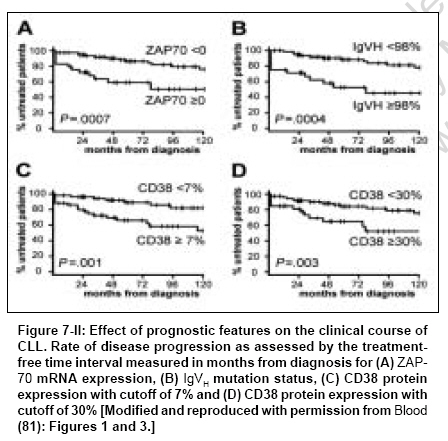
Indian Journal of Cancer, Vol. 44, No. 2, April-June, 2007, pp. 72-86 Review Article Clinical implication of genome-wide profiling in diffuse large B-cell lymphoma and other subtypes of B-cell lymphoma Iqbal Javeed, Joshi Shantaram, Patel Kavita N, Javed Sofi I, Kucuk Can, Aabida Afeera, d'Amore Francesco, Fu Kai Department of Pathology and Microbiology, Genetics Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, NE Code Number: cn07012 Abstract The differentiation of lymphoid cells is tightly regulated by transcription factors at various stages during their development. During the maturation processes, different genomic alterations or aberrations such as chromosomal translocation, mutation and deletions may occur that can eventually result in distinct biological and clinical tumors. The different differentiation stages create heterogeneity in lymphoid malignancies, which can complicate the diagnosis. The initial diagnostic scheme for lymphoid diseases was coined by Rappaport followed by Revised European and American Classification of Lymphoid Neoplasms (REAL) and World Health Organization (WHO) classifications. These classification methods were based on histological, immunophenotypic and cytogenetic markers and widely accepted by pathologists and oncologists worldwide. During last several decades, great progress has been made in understanding the etiology, pathogenesis and molecular biology of malignant lymphoma. However, detailed knowledge in the molecular mechanism of lymphomagenesis is largely unknown. New therapeutic protocols based on the new classification have been on clinical trials, but with little success. Therefore, it is imperative to understand the basic biology of the tumor at molecular level. One important approach will be to measure the activity of the tumor genome and this can partly be achieved by the measurement of whole cellular mRNA. One of the key technologies to perform a high-throughput analysis is DNA microarray technology. The genome-wide transcriptional measurement, also called gene expression profile (GEP) can accurately define the biological phenotype of the tumor. In this review, important discoveries made by genome-wide GEP in understanding the biology of lymphoma and additionally the diagnostic and prognostic value of microarrays are discussed.Keywords: Classification, diffuse large B-cell lymphomas, gene expression profiling, prognosis, targeted therapy Introduction There has been a significant advance in our understanding of the pathogenesis and biology of lymphoid malignancies. [1] Translocation breakpoints associated with many types of lymphomas [1],[2],[3] have been cloned and their mechanisms of action are being elucidated. Many molecular abnormalities or markers with significant diagnostic or prognostic importance have been identified, such as the activation of oncogenes through specific translocations and other mechanisms, [4],[5],[6],[7],[8],[9] the inactivation of tumor suppressor genes [10],[11],[12],[13],[14],[15],[16],[17] and abnormalities in the apoptotic pathways. [18],[19],[20],[21],[22],[23],[24] Traditionally, the investigation of these parameters has been performed individually, rather than globally. Recognizing the potentials of DNA microarray technology when coupled with a large tumor/clinical resource, a number of groups have explored the use of gene expression profiling in lymphoma diagnosis, classification and outcome prediction. The application of this technology has, in the past few years, led to the discovery of unique gene expression signatures for the major groups of B-cell non-Hodgkin's lymphoma (NHL), the discovery of several novel subtypes of diffuse large B-cell lymphoma (DLBCL), [25],[26],[27],[28],[29] the construction of molecular prognosticators [26],[27],[30] and insight into the molecular mechanisms that determine the behavior of a tumor. [23],[31],[32] Herein, we will briefly review recent advances in the molecular diagnosis of DLBCL and other lymphoma types using gene expression profiling as a tool. It does not intend to be a comprehensive review of DLBCL, but rather it will illustrate examples of how gene expression profiling has been used in disease classification and outcome prediction.Principle of Gene Array The development of current microarray technology for gene expression profile (GEP) can be attributed to some of the pioneering studies of the last decade and reviewed previously. [33],[34] GEP examines expression for thousands of transcripts (mRNA) simultaneously in a single experiment. Known oligonucleotides (DNA probes) or complementary DNA (cDNA) PCR products are immobilized on a chemically active surface attached to a solid support of glass, also called gene chips or DNA microarrays. The probes can also be pre-synthesized as single-stranded oligonucleotides of 50-60 bases or in situ synthesized on surface. Gene chips can hold 10,000-25,000 different genes and are available commercially or custom made at core facilities in universities or research centers. The assay of GEP starts with total RNA isolation from fresh or frozen tissues followed by synthesis of cDNAs/cRNAs, which are labeled with fluorescent dyes, such as Cy3, Cy5 or phycoerythrin-conjugated streptavidin [Figure - 1]. The labeled cRNAs/cDNAs are then hybridized on gene chips to measure the abundance of the transcripts in cells. This is accomplished by scanning the chips through confocal-assisted lasers and then a thorough analysis of the images with various computational programs. The GEP data are quantitative and highly reproducible, therefore mathematically tractable. Several statistical and mathematical models have been developed for data analysis to generate a meaningful hypothesis. [35],[36]Unique Gene Expression Dignatures in Major Types of B-cell NHLs Diffuse Large B-cell Lymphoma Diffuse large B-cell lymphoma Accounts for 35-40% of total non-Hodgkin malignant lymphomas (NHLs) and is the major diagnostic category of NHL. [37],[38],[39] The four major morphological variants according to the WHO classification of DLBCL (centroblastic, immunoblastic, T-cell/histiocyte rich and anaplastic) proved to have neither biological nor clinical significance. Half of the patients diagnosed with DLBCL can be cured and the rest of them succumb to the disease, suggesting an enormous clinical heterogeneity. The first clue to the puzzle of the heterogeneous nature of this disease came through a GEP study, which revealed at least two distinct subgroups of the disease [25],[27] [Figure - 2]. The diversity in this malignant lymphoma appears to be associated with the different differentiation stages and proliferation rates of B cells. Some of the biological insights from GEP studies in DLBCL have been reviewed by Fu et al ., Iqbal et al . [40],[41] and Dybkaer et al . [42] These studies identified three distinct subgroups of DLBCL, one of which demonstrated a germinal center B-cell (GCB) like gene expression pattern. [25],[27] The genes overexpressed in this subgroup include GC markers such as CD10, BCL6, A-MYB and OGG1 . It also showed somatic hypermutation in immunoglobulin (Ig) gene owing to the germinal center reaction. [43] This subgroup also showed a frequent genomic translocation t(14;18) involving the BCL2 gene. [44],[45] The second subgroup revealed activated B-cell (ABC) like pattern and overexpressed genes involved in mitogenesis such as cyclin D2, FOX-p1, CD44 and IRF-4 . This subgroup lacks t(14;18) and Ig hypermutation; however, it is highly associated with amplification of 18q21 region. [46] Patients with GCB-like DLBCL had a significantly better overall survival than those with ABC-like DLBCL. The third subgroup identified as primary mediastinal large B-cell lymphoma (PMBL) is discussed in the next section. The fourth subgroup of patients, accounting for 20% of the total cases, showed neither GCB-like nor ABC-like gene expression pattern. They shared a similar risk index with the ABC subtype and are regarded as 'unclassifiable'. The association of DLBCL heterogeneity with the cell of origin hypothesis was further complimented by an independent study. [30] The authors used a supervised approach to guide the discovery process in analyzing GEP for patients with cured versus fatal or refractory disease groups and confirmed DLBCL subgroups. Primary mediastinal Large B-cell lymphoma: A subgroup of DLBCL related to Hodgkin's lymphoma Primary mediastinal large B-cell lymphoma is characterized by its anatomic location, clinical presentation, histological and immunophenotypic features. The tumor cells express CD30 and B-cell markers (CD20, CD79a, PAX5, BOB.1, OCT-2 and PU.1) but not surface Ig and CD15. Majority of the cases with PMBL demonstrated Ig somatic hypermutation and proved to be of post-germinal center cell origin. Although aggressive chemotherapy with MACOP has shown long-term disease-free survival in majority of the patients, a minority of the patients finally die of the disease, indicating the heterogeneity by histological and clinical classification. GEP studies by Rosenwald et al . and Savage et al . showed a unique gene expression signature for PMBL, [26],[29] which included downregulation of several B-cell receptor components and molecules involved in its downstream signaling (BLK, BLNK, PKC βI, NFATc and CD22). Several functional groups of genes were upregulated such as the cytokine pathway components (IL13R α1, JAK2, STAT1, NF-IL3, RANTES and IP10), the tumor necrosis factor (TNF) family members and induced proteins [OX40ligand (TNFSF4), CD95, TRAF1 and TNFa induced protein 3] and the extra-cellular matrix element (TIMP1) [Figure - 3]. Another significant observation of this study is the similarity of the gene expression pattern to the classic HL suggesting that both PMBL and HL cells may originate from a thymic B-cell. Several genes expressed in PMBL are characteristically expressed in Hodgkin Reed-Sternberg cells such as CD30, IL13Rα and TARC. Clinical features also showed some similarity between PMBL and HL, like the occurrence in young woman. The patients with PMBL are younger than DLBCL with average age of 32 versus 64 showing better overall five-year survival (64% versus 30%). Mantle cell lymphoma Mantle cell lymphoma (MCL; 3-10% of NHL) is the malignant counterpart of mantle zone B-cells and is considered a distinct type of mature B-cell lymphoma with aggressive clinical behavior. [47] The MCL cells bear surface immunoglobulin (IgM and IgD) and are often CD5+. This entity of lymphoma has a unique feature of t(11;14)(q23;q32) translocation, which results in upregulation of cyclin D1 gene in the tumor cells. The genomic deletions spanning the ataxia telangiectasia mutated (ATM) gene have been observed at the highest frequency in MCL. [48] The survival of MCL patients is highly variable and no effective therapy has been developed. Rosenwald et al . successfully established the MCL gene signature in a large series (101 cases), with which 98% of cyclin D1 positive MCLs were correctly diagnosed. [28] Seven of nine cases morphologically typical for MCL but negative for cyclin D1 expression showed the same gene expression signature, thus providing molecular evidence for the presence of cyclin D1 negative MCL. This was further confirmed by a subsequent study of six cases of cyclin D1 MCL suggesting that cyclin D1 and D3 can substitute the role of cyclin D1 in this subgroup. The precise measurement of tumor cell proliferation, provided by the expression of proliferation signature genes, identified patient subsets that differed by more than 5 years in median survival [Figure - 4]. Differences in cyclin D1 mRNA abundance synergized with INK4a/ARF locus deletions to dictate tumor proliferation rate and survival. The proliferation gene expression signature functioned as a quantitative integrator of multiple oncogenic aberrations. Some additional GEP studies with fewer cases reported abnormalities of the apoptotic pathway in MCL [49] such as downregulation of FADD (FAS-associated via death domain), DAXX (death-associated protein 6), RAIDD and caspase 2. Follicular lymphoma Follicular lymphoma (FL; ~22% of all cases of NHL) is typically an indolent disease, but essentially incurable. FL, which is a GC-derived B-cell malignancy, has the tendency to transform into morphologic and clinical DLBCL, where t(14;18)(q32;q21) is a common genetic alteration (90% of cases). [50] This translocation leads to the overexpression of the anti-apoptotic protein BCL2, hence resulting in accumulation of follicle center cells by virtue of prolonged cell survival. There is an outgrowth of a more malignant subclone in 25-60% of FLs, with a rapidly progressive clinical course and short survival time. This transformation is accompanied by genetic changes such as MYC gene rearrangement, p53 mutation, [16] mutations in the 5´ untranslated region of the BCL6 gene, [51] mutations of the translocated BCL2 gene [52] or inactivation of P15 or P16 by deletions, mutations and hypermethylations. [14],[53],[54] These secondary genetic aberrations are not present in all transformed lymphomas suggesting more than one mechanism of transformation. This idea is supported by a GEP study where two different GEPs were observed to be associated with the transformation of FL to DLBCL: one showed increased expression of the oncogene MYC and the genes regulated by MYC, whereas the other showed decreased expression of the same genes. The study also revealed that transformed FL has different GEPs from de novo DLBCL. The array comparative genomic hybridization (CGH) data showed that FL transformation is associated with genomic imbalances such as amplification of 2p16 (including the REL/BCL11A loci), 3q27-q29 (containing the BCL6 locus), 18q21 (including the BCL2 locus) as well as deletion of 17p13 (including the p53 locus). None of the transformed cases had elevated copy numbers of the MYC gene locus on 8q24 according to the array CGH. However, several of the MYC target genes such as NME1 in 17q21, JTV1 in 7p22, CYP51 and CUTL1 in 7q22, CDK2 and CDK4 in 12q and AHCY in 20q11 were located in aberrant genomic areas of amplification or loss accompanying the transformation from FL to DLBCL. In another study, several members of the RAS family and p38MAPK pathway were shown to have high expression levels in transformed FL. The authors showed that inhibition of p38MAPK blocked the growth of t(14;18) positive cell lines and inactivation of p38MAPK inhibited tumor growth in NOD-SCID mice by inducing apoptosis, thereby suggesting that pharmacologic targeting of p38MAPK may be an effective strategy for developing new therapies against transformed FL. Burkitt's lymphoma Burkitt's lymphoma (BL) is an aggressive B-cell lymphoma that accounts for 30-50% of lymphomas in children but only 1-2% of lymphomas in adults. [55] It is rapidly fatal if untreated, but curable with intensive chemotherapy. BL is not, however, curable by the treatment for DLBCL, which typically consists of cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP), with the monoclonal anti-B-cell antibody rituximab. Diagnostic accuracy is, therefore, essential to prevent the undertreatment or overtreatment of patients. BL is characterized by a translocation involving the c-MYC gene and one of the loci encoding the Ig heavy or light chains. These features are distinctive, but they overlap with morphologic and genetic attributes of DLBCL. Recently, two large-scale studies investigated if GEP could reliably distinguish between these two entities. In these two reports, Hummel et al. and Dave et al. studied a total of 523 cases of aggressive lymphomas. [56],[57] Eighty-one cases were classified as classical BL or atypical BL by expert hematopathologists based on established WHO criteria. Among these, 72 cases (90%) exhibited a characteristic molecular signature of BL [Figure - 5]. Others exhibited a molecular signature of DLBCL or an undefined group intermediate between DLBCL and BL. Thus, nine patients might have been overtreated for what was really DLBCL or non-BL. Moreover, both studies showed that gene expression seems to outperform the expert hematopathologists since approximately 4.6% of the cases diagnosed as DLBCL or 21% of the cases diagnosed as unclassifiable high-grade B-cell lymphomas by experts also exhibited a characteristic molecular signature of BL [Figure - 5]. This finding suggests that up to 5% of patients with DLBCL whose diagnoses were based on the current histological and immunophenotypic criteria might have been undertreated. Hummel et al. also identified a distinctive BL signature, which consisted of 58 genes, including several target genes of the nuclear factor-kB (NF-kB) pathway such as BCL2A1, FLIP, CD44, NFKBIA, BCL3 and STAT3. These genes were expressed at lower levels in BL cases than in DLBCL cases. Moreover, Hummel et al . examined the global genetic abnormalities using FISH and array-based CGH. They showed that 88% of the cases with Burkitt's signature had translocations between c-myc and one of the Ig loci in the background of a karyotype in which few other abnormalities were present. Hummel et al. demonstrated that three main cytogenetic groups could be distinguished within the mature aggressive B-cell lymphomas [Table - 1]. The BL group predominantly consisted of myc-simple lymphomas; the non-BL group predominantly consisted of myc-negative lymphomas. In contrast, the intermediate group (intermediate between BL and DLBCL) contained most of the myc-complex cases, occasional myc-simple and several myc-negative cases. They further showed that a c-MYC rearrangement in tumors without Burkitt's signature was an important predictor of poor outcomes. [56] Other lymphomas and leukemias Splenic Marginal Zone Lymphoma (MZL): A recent study by Conconi et al . reported that splenic MZL could be successfully distinguished by GEP from small lymphocytic lymphoma (SLL) and MCL using the 44-gene predictor. [58] SLL discriminating genes were related to cell adhesion (P- and L-selectin, integrin 5, LAMR1, COL4A4, COL18A1, CCR2 and MIF), angiogenesis (angiopoietin-like 3, FIGF), cell fate (notch 4, IL4R and TNFR17) and anti-apoptosis (BCL2, survivin and TNFR10). [58] MCL discriminating genes were related to three functions: cell proliferation (PCNA, CDK4, cyclins F, DNMT1, CDC46, MYBL2 and topoisomerase A2), gene transcription (TFAP2A, TCF3, SRF, NFE2L3 and SMARCA4) and drug resistance [GST pi and two MDR/ATP-binding cassette (ABC) membrane proteins (ABCG2 and ABCC5)]. Splenic MZL discriminating genes regrouped 83 genes from two clusters. One of the two clusters was specifically associated with the MZL signature, whatever the site of the biopsy (spleen, node or blood). These genes were involved in intracellular signaling (AKT1 and AGER) or transcription (TFCP2 and MXI1). Several S100A proteins [S100 A8 and A9 (caligranulin a and b)] positively discriminated the MZL samples. The second splenic MZL signature corresponded to the 'sPLEEN' cluster and was related to genes expressed by cells other than lymphoma B cells such as red cells (e.g. hemoglobin zeta) and T cells, considered as contaminant cells interfering with the MZL signature. Primary cutaneous large B-cell lymphomas: Primary cutaneous large B-cell lymphomas (PCLBCL) are a heterogeneous group of B-cell neoplasms, with two distinct subtypes, primary cutaneous follicle center cell lymphoma (PCFCCL) and primary cutaneous large B-cell lymphoma of the leg (PCLBL-leg). Patients with PCLBL-leg usually present with solid or multiple nodular lesions on legs, which are histologically characterized by tumorous infiltrates of large centroblasts and immunoblasts with large round nuclei. PCFCCL demonstrated more localized disease with histological features showing a spectrum of centrocytes and centroblasts in diffuse or follicular patterns. [59] PCFCCL showed more favorable prognosis (five-year survival of 95%) than PCLBL-leg (five-year survival of 50%). GEP of PCLBCL demonstrated the distinction between these two subtypes of neoplasm at molecular levels. [60] Eight of 21 cases were classified into PCFCCL and the rest were classified as PCLBL-leg by hierarchical clustering based on B-cell expression signature. PCLBL-leg showed higher expression of genes associated with cell proliferation, which included c-MYC, PIM1, PIM2, MUM1/IRF4 and OCT2. MUM1 was constantly detected by immunohistochemical staining in PCLBL-leg but not in PCFCCL. PCFCCL showed gene expression similar to germinal center cell-like DLBCL, whereas PCLBL-leg exhibited a signature similar to ABC-DLBCL. Chronic lymphocytic leukemia: B-cell chronic lymphocytic leukemia (B-CLL) is a heterogeneous disease of CD5/CD19/CD23 positive B cells with an extremely variable clinical course, i.e. from indolent to very aggressive behavior. Klein et al. have shown that GEP of B-CLL is similar to that of memory B cells as compared to naοve B cells or CD5 positive B cells or centroblasts/centrocytes of the germinal centers. Although alkylating agents and nucleoside analogs have been used to treat CLL, the overall survival time has remained at approximately 6 years. [61],[62] For the last few years, several investigators have reported the GEPs of CLL cells and their relationship to biologic properties and clinical outcomes. Molecular analyses of peripheral blood CLL cells have not revealed any significant gene expression pattern that has biological and clinical significance for CLL. This is in contrast to the findings in a number of other hematological malignancies with variable clinical outcomes such as DLBCL, [41] multiple myeloma (MM) [63] and acute lymphoblastic leukemia (ALL) where GEPs correlated with relevant prognostic and biological subgroups. [64] Many of the CLL GEP studies have focused on correlating the gene expression signature with particular known and accepted prognostic markers. Rosenwald et al. and others have reported that CLL cells have a uniform gene expression signature irrespective of Ig mutational status and identified several genes that correlate with Ig mutational status. [65],[66] Recently, we have reported that the expression of certain genes is associated with subgroups of CLL with different chromosomal abnormalities. For example, overexpression of CDC2, CD25A and Cyclin-C is associated with the 11q deleted subgroup and overexpression of HRK gene with the trisomy 12 subgroup. [67] Furthermore, Haslinger et al. and our group have shown that significantly differentially expressed genes are located in the corresponding abnormal chromosomal regions indicating a gene dosage effect. [68],[69] Few studies have reported that CLL cells express cell cycle/apoptosis regulatory genes that are specific for activated cells in G 1 -S phase of the cell cycle [70] and a correlation exists between CLL patients' survival and the expression of genes encoding cell adhesion molecules like L-selectin and integrin β-2 or factors that induce these molecules, such as IL-1β, IL-8 and EGR 1 (early growth response protein 1). [71] Guidance to Molecular Prognosticators and Theaurepetic Target Discovery The DNA microarray data in combination with clinical data of cancer specimen can generate meaningful biological insights into the tumor. The biological phenotype can also be associated with gene expression signatures, which can serve as quantitative measures of tumor behavior. This method of analysis, also called 'supervised', uses statistical methods to find correlation between gene expression data and external parameters (clinical, pathological or molecular). This leads to sets of genes that are discriminatory between different subgroups of lymphoma. Often, the use of statistical models can also indiscriminate the choice of genes, due to overfitting of some genes that are particular to the data sets on which they are based. [72] Therefore, microarray data sets are divided into 'training' and 'validating/testing' sets to generate more meaningful gene models. However, models with more biological insights relating to the external clinical behaviors are more reliable for the reason that transcriptional co-regulation of genes in a similar biological process is well coordinated. This principal has been used to derive survival predictors in a number of lymphoid malignancies as described in the following. Diffuse large B-cell lymphoma The three subgroups of DLBCL differ in their clinical behavior, which can also be related to the expression of different groups of genes. This distinction is independent of the international prognostic index (IPI) and therefore not a surrogate for clinical parameters. Four different signatures were defined based on their associations with the length of survival [Table - 1]. A multivariate model formed from these signatures was able to divide the cases into four quartiles, with five-year survival rates of 73%, 71%, 34% and 15%, respectively. [27] High expression of the three genes representing the GCB signature (BCL6, GCET1 and GCET2) correlated with a good prognosis. Overexpression of the three genes representing the proliferation signature (MYC, E21G3 and NPM3) correlated with a poor prognosis. Another significant difference between the DLBCL subgroups is the activation of the NF-kB pathway in the ABC [23] and PMBL subgroup. [73] Inhibition of the NF-kB signaling pathway induces apoptosis in ABC cell lines but not in GCB cell lines. This observation indicated different pathogenetic mechanisms in the two DLBCL subgroups and the possibility of selective therapy in subgroups of DLBCL. Recently, Monti et al. identified various biological parameters associated with DLBCL through robust computational models called consensus clusters (CC). [74] They identified three robust subsets of DLBCL, including oxidative/phosphorylation (OxPhos), B-cell receptor/proliferation (BCR/Proliferation) and host response (HR). The OxPhos signature includes genes involved in oxidative phosphorylation, suppression of apoptosis and mitochondrial and proteasome function. The BCR/proliferation subset is enriched for genes involved in cell-cycle regulation, DNA repair, cell division and B-cell-receptor (BCR) signaling. The third subset, HR, is enriched for genes involved in T-cell-receptor signaling (ZAP70 and LAT), natural killer (NK) cell activation (NKp30), complement cascade, cytokine signaling (IRF1-7), dendritic cell maturation (LAG3/CD223) and T-cell adhesion and chemotaxis (LFA1 and CXCR6). In contrast to the cells of origin model, the CC model demonstrated no correlation with tumor responsiveness to conventional therapy; however, it can be used for new potential targets for therapy. Primary mediastinal large B-cell lymphoma Two independent GEP studies were able to clarify the distinct category of PMBL from other subgroups of DLBCL. [26],[29] The two studies complemented each other and six of the 20 genes [Figure - 3] included in the final predictor defined by Rosenwald et al. are in common with the predictor defined by Savage et al. Both studies were also able to find a relationship of PMBL with HL and that several genes (MAL and IL41) are expressed by HL cells. PMBL cases compared to other DLBCLs, expressed increased levels of NFaB targets that promote cell survival and favor antiapoptotic TNF signaling. In contrast, ABC-like DLBCLs had a more restricted, potentially developmentally regulated NFaB target gene signature. In addition, NFaB activation was not associated with amplification of the cREL locus, suggesting alternative pathogenetic mechanisms. PMBLs have a chromosomal gain or amplification of 9p24 cytoband in 40-50% of patients, which leads to overexpression of PDL1/PDL2 and JAK2. [26] The overexpression of PDL1 and PDL2 may inhibit the tumor specific T-cell response to the lymphoma and therefore can be an attractive target specific for PMBL. Mantle cell lymphoma Supervised approach in MCL resulted in identification of a 'proliferation signature', which was a better predictor of survival than the morphological categorization (P = 5.7 x 10-3). The proliferation signature included genes involved in cell-cycle progression and DNA synthesis, but not c-MYC, therefore reflective of the tumor proliferation rate [Figure - 4]. Patients with different predictor scores (average of 20 proliferation genes) differed by more than 5 years in medial survival. The quartile of patients with the highest level of proliferation-signature expression had a median survival of 0.83 years, whereas the quartile with the lowest level of expression had a median survival of 6.71 years. Deletion of the INK4a/ARF locus encoding p16 INK4a and p14 ARF was detected in 21% of the cases and these cases had a relatively poor prognosis. The tumor suppressor protein p16 INK4a controls transition from G1 to S in the cell cycle. [75] The p14 ARF protein sequesters MDM2, which is a negative regulator of p53, thereby allowing p53 to mediate apoptosis and cell cycle arrest in the proper setting. [76] Noteworthy was also the overexpression of BMI-1, a transcription repressor of p16INK4a and p14ARF , [76] seen only in the MCL cases without deleted INK4a/ARF locus. [77] Thus, from therapeutic point of view, targeted therapy to lower down the proliferation rate in MCL can be of clinical benefit to the patients. Follicular lymphoma Dave et al. developed a molecular predictor of survival and identified biologic determinants of the clinical heterogeneity of FL. [32] The analysis was supervised on the basis of length of survival and clusters of genes with highly correlated expression patterns that were grouped into survival-associated signatures. They identified two signatures of coordinately regulated genes that together represented the best predictor of survival. When patients were grouped into quartiles on the basis of survival-predictor scores, patients in the top quartile had a median survival of 13.6 years, whereas those in the bottom quartile had a median survival of only 3.9 years [Figure - 6]. Interestingly, the genes that best defined the prognostic signatures in FL were expressed primarily by T cells, macrophages or dendritic cells, but not by the tumor cells themselves. This finding strongly indicates that the aggressiveness of the disease is mainly determined by the cellular microenvironment of the lymphoma and not by obvious differences in GEP of the malignant cells themselves, although the cellular microenvironment may well be influenced in important ways by the lymphoma cells. Thus, from the molecular standpoint, FL is clearly different from DLBCL and MCL, in which the prognostic signatures are based largely on genes expressed by the lymphoma cells. Burkitt's lymphoma Dave et al. used hierarchical clustering to organize Burkitt's signature genes and revealed four prominent gene groups. [57] The c-myc protein and its target genes constituted one group, which was highly expressed in BL as compared to DLBCL. The second group included genes that were expressed in normal germinal-center B cells. [56] These genes were highly expressed in BL compared to GEC-DLBCL and termed as the 'BL-high' signature [Figure - 5]. The third group included major-histocompatibility-complex (MHC) class-I genes and the fourth group included NF-kB target genes. The genes in the third and fourth groups were expressed at lower levels in BL than in DLBCL. Hence, Burkitt's lymphoma was readily distinguished from DLBCL by the high level of expression of c-myc target genes, the expression of a subgroup of germinal-center B-cell genes and the low level of expression of MCH class-I genes and NF-kB target genes. Among the 28 patients with a molecular diagnosis of Burkitt's lymphoma, the overall survival was superior among those who had received intensive chemotherapy regimens instead of lower-dose regimens. Hummel et al . also identified a distinctive BL signature, which consisted of 58 genes, including several target genes of the NF-kB pathway such as BCL2A1, FLIP, CD44, NFKBIA, BCL3 and STAT3. These genes were expressed at lower levels in BL cases than in cases of DLBCL. Chronic lymphocytic leukemia Prognostic indicators such as IgV H gene mutational status, cytogenetic abnormalities, CD38 expression and ZAP-70 expression have been shown to correlate with clinical outcomes. Subgroups of CLL with unmutated IgV H gene have poor prognosis, whereas the CLL subgroups with mutated IgV H gene have a favorable prognosis [65] [Figure - 7a, b]I. CD38 expression is correlated with poor prognosis while better prognosis is associated with decreased expression of CD38 on CLL cells. [65],[78] Several investigators have shown that patients with chromosomal abnormalities of 13q deletion and normal karyotypes have a better clinical outcome and long-term survival compared to patients with 17p deletion, 11q deletion and trisomy 12 and correlated this with a specific gene signature. [67],[79] Expression of ZAP-70 gene, a member of the Syk/Zap-70 protein kinase family, has been shown to be another potential prognostic marker in CLL. [80],[81] Patients who had greater expression of ZAP-70 had a poor prognosis compared to patients who had lower expression of ZAP-70 gene. Thus, based on availability and reliability of these prognostic factors, we are now in a better position to predict progress in many CLL patients [Figure - 7a, b]II. Genomic expression profiling using microRNAs, a class of small, non-coding but functional RNAs has been proven to be a very useful tool in analyzing gene expression patterns between normal and abnormal cells. Recently, analyses of CLL cells for the microRNAs have revealed useful information in predicting prognosis and disease progression [82],[83],[84],[85] and revealed a distinct signature in CLL prognostic subgroups using human precursor and mature microRNA oligonucleotides as probes. Thus, genomic expression profiling has the potential to be a reliable method to predict outcome in CLL cells at the time of diagnosis. Further studies from several different laboratories will be required to support this notion. Taken together, gene expression patterns based on known prognostic markers have yielded useful results including identification of key genes involved in CLL biology leading clinical behavior. Thus, genomic profiling is becoming more and more useful. Challenges and Key Issues in Translating of GEP Findings into Clinical Diagnosis Studies in the past several years have shown quite clearly that GEP has made a significant impact on the molecular diagnostics of lymphomas. The application of this technology has led to the discovery of novel types of lymphoma, molecular prognosticators, as well as insights into the molecular mechanisms that determine the pathogenesis and the behavior of a tumor. With the continued application of this and other novel technologies, we expect lymphoma diagnosis, classification and survival prediction to keep evolving. Molecular diagnostics, in the future, will be expected to provide more specific information concerning the pathobiology of each tumor sample, including the key aberrant pathways. Ideally, therapy will be based on the information, aiming at the most effective mechanism-based treatment with the least toxicity. DNA microarray assays require well-preserved RNA, which is generally obtained from fresh or snap-frozen tissues. Thus, for routine use of microarray assays in clinical laboratories, it would require a widespread acceptance of submitting tissue biopsies in the fresh state with a representative sample reserved for microarray analysis. This may pose as a considerable obstacle since it may be difficult to change the traditional pattern of the tissue handling in many institutions and the fresh tissue may not be available due to financial organizational or infrastructure limitations. While the use of RNA from paraffin tissues for microarray studies has been reported, it is not known if this technology will be sufficiently robust for clinical applications. It is, therefore, important to adapt the established gene expression signatures to a platform suitable for routine tissue processing in the clinical laboratories. One strategy for translating microarray profiles into clinical practice is to first identify small, diagnostic gene expression signatures with array studies and then to validate the clinical utility of these genes either retrospectively or prospectively with the use of simple, robust, conventional assays such as quantitative real time polymerase chain reaction (QRT-PCR) or immunohistochemical staining. While current gene expression signatures frequently contain a substantial number of genes, it has been shown in a number of studies, that this number can be substantially reduced without significantly compromising the discriminatory power of the original signature. Quantitative real-time PCR The quantitation of well-preserved mRNA by RT-PCR is currently well established and the application of this technology to RNA from paraffin-embedded tissue has also been reported by several groups. [86],[87],[88],[89],[90],[91] A recent comprehensive study by Cronin et al. has clearly demonstrated that it is a viable approach for archival tissues and can be performed as a moderately high throughput assay. [92] The expression of a variety of genes measured either individually or in large data sets derived from microarrays had been reported to predict survival in DLBCL. Among the long list of these genes, Lossos et al. measured the expression of 36 genes in independent samples of DLBCL from 66 patients by QRT-PCR analysis and related the expression levels of these genes to overall survival. [93] In a univariate analysis, genes were ranked on the basis of their ability to predict patient survival. A small group of six genes that were the strongest predictors, including LMO2, BCL6, FN1, CCND2, SCYA3 and BCL2, was identified. The authors then developed a multivariate model that was based on the expression of these six genes and the model was further validated in two independent microarray data sets. It has been further shown that the model was independent of the IPI values and added to its predictive power. LMO2 and BCL6 are in the germinal-center B-cell signature, while BCL2, CCND2 and SCYA are highly expressed in the ABC-DLBCL and FN1 in the lymph-node signature. Therefore, the six-gene predictor basically reflects the GCB-/ABC-DLBCL distinction and stromal signature. The study demonstrated that a small number of genes could capture a significant amount of information from gene expression profiling and indicated that an RT-PCR platform can be employed as an alternative platform. Further refinement in gene selection may allow the use of the QRT-PCR as a technically simple and robust platform for routine clinical applications. Immunohistochemical analysis For a gene expression parameter to be successfully transferred to an immunohistochemical platform, the following conditions must be met: (i) a correlation between the level of the transcript and protein expression exists; (ii) a suitable antibody for immunohistochemistry is available; (iii) the epitope recognized by the antibody is stable and retrievable for antibody binding; and (iv) the reaction is robust and readily quantifiable. In a recent study from our group, a series of antibodies against CD10, BCL6, MUM1/IRF-4, FOXP1, cyclin D2 and BCL2 were used on 152 cases of DLBCL, 142 of which had been successfully evaluated by cDNA microarray. [94] Expression of BCL6 or CD10 was associated with better overall survival, whereas expression of MUM1/IRF-4 or cyclin D2 was associated with worse overall survival. Cases of DLBCL were then subclassified using CD10, BCL6 and IRF-4/MUM1 expression [Figure - 6], 64 cases were considered as GCB-DLBCL and 88 cases as non-GCB-DLBCL. The five-year overall survival for the GCB group was 76% compared with only 34% for the non-GCB group (P< 0.001). The results are comparable to those reported using the cDNA microarray. Similar results have been obtained from other groups as well. Additional antibodies will likely be identified or developed in the future, which may markedly improve the predictive power of the immunostaining panel in the diagnosis and prognosis of lymphomas. We have cloned and sequenced the full-length cDNA of two of the best GCB markers, [95],[96] GCET1 and GCET2/HGAL. A rabbit polyclonal antipeptide antibody has been prepared for GCET2 while antibodies to GCET1 are being developed. A monoclonal antibody against the GCET2/HGAL protein has also been developed. Among the 718 lymphomas tested by immunohistochemistry, staining for GCET2/HGAL protein was present in 97% (103/107) of follicular lymphoma, 100% (40/40) of BL, 87% (7/8) of mediastinal large B-cell lymphoma and 70% (103/151) of DLBCL. [97] While experienced pathologists can provide reasonably accurate and reproducible scoring of immunohistochemical stains, it is still subjective and is a significant source of variance. This variance can be markedly reduced if an accurate, automated, quantitative image processing system is available. Novel technologies that will significantly enhance our ability to quantitate immunohistochemistry will be of great interest as quantitation may be necessary for certain parameters. Gene Expression Profiling from a Clinician's Perspective Since the late 1970s, combination chemotherapy (e.g. CHOP, cyclophosphamide, hydroxydaunomycin, vincristine and prednisone) has been responsible for a significant improvement in the prognosis of aggressive NHL. [98] Subsequently, regimens more complex than CHOP, based on single-institutional phase-II trials, seemed to further improve long-term survival rates from a previous 30-35% to 55-65%. [99],[100],[101] However, when compared in large randomized multi-center trials, most of these regimens did not prove to be more effective than the standard-dose CHOP, [102] suggesting that the high phase-II cure rates were probably a result of patient selection. This observation emphasized the need to compare therapeutic regimens in patients with comparable risk profiles aiming at tailoring treatment strategies to different risk cohorts. As a result of those efforts, an easily reproducible and therefore widely accepted prognostic model, the IPI, was developed in the early 1990s. It contains pretreatment clinical features independently predictive for overall and relapse-free survival. [38] The clinical factors identified by the IPI reflect three basic features: (i) the tumor's growth and invasive potential (serum-lactate dehydrogenase, clinical stage and number of extranodal sites), (ii) the patient's response to the tumor (performance status) and (iii) the patient's general condition and ability to tolerate intensive chemotherapy (age and performance status). The number of adverse prognostic factors present in a given patient prior to treatment start provides a reproducible risk prediction (low, low-intermediate, high-intermediate and high risk) by which patient cohorts with similar risk profiles could be analyzed and compared. After the advent of a new and more biologically oriented lymphoma classification according to the REAL/WHO criteria, [39] some shortcomings of the IPI have been reported, e.g. the fact that it is a better prognostic discriminator for DLBCL than for other NHL subtypes such as FL or peripheral T-cell lymphoma (PTCL). [44] However, for routine clinical purposes, the IPI is currently still one of the most useful and most widely adopted prognosticators in NHL patients. Today, new biotechnological tools have helped clarifying the striking clinical and genetic heterogeneity found within some of the major lymphoma entities recognized by the 2001 WHO classification. These tools are also suitable for the identification of pathogenetic mechanisms and subtype-specific molecular targets exploitable for therapeutic intervention. In particular, cDNA microarray for GEP has been among the most data-generating approaches so far. By an unsupervised approach, highly reproducible tumor-specific transcriptional gene signatures have made it possible to identify homogeneous subgroups of tumors with related signatures [25],[26],[27],[28] and by a supervised approach, to correlate specific signatures to distinct survival patterns. [41] In terms of outcome prediction, GEP also seems to provide additional prognostic information independent from the IPI. So far, molecular predictive models have mainly been developed on patients treated before the advent of immunotherapy. The molecular predictors of clinical outcome need, therefore, to be validated also in the setting of modern combined treatment modalities (e.g. immunochemotherapy or radioimmunochemotherapy). However, already now, the consistent findings obtained by the large number of published microarray-based GEP studies allow to rationally designing clinical trials in which treatment cohorts are stratified according to molecular profiling criteria. This approach will test the prognostic impact of GEP in a prospective, even randomized manner and may lead to the recognition of genes that critically influence the response to investigational drugs. The lymphoid malignancies that, on the basis of available data, seem to be an appropriate initial target group for such an approach, are entities such as DLBCL, MCL and CLL. Conclusion With knowledge generated from microarray and other studies on various forms of malignant lymphoid disorders, the diagnosis and classification of lymphomas should include measurement of parameters most relevant for determining the biologic and clinical behavior of each tumor so that patients can be segregated into prognostically distinct subsets. Gene expression profiling studies of lymphomas have included thousand of genes but as demonstrated in many of the papers in this review, the expression patterns of a limited number of genes are often sufficient to characterize specific subgroups of lymphomas, a feature that is important when aiming at developing arrays to be used in routine diagnosis and prognosis. Researchers have used different microarray platforms in their investigations and comparison of data across these platforms is a major challenge. An example of cross-platform comparison is the recent study by Wright et al. where two sets of data on DLBCL were compared. The GCB and ABC subgroups of DLBCL defined on the Lymphochip could be identified from an Affymetrix data set with a new statistical model and the two subgroups could be shown to maintain their specific correlation with survival in a platform-independent manner. The most promising potential of gene expression profiling studies is the ability to define new therapeutic targets. One example of this is illustrated by the profiling studies of DLBCL where the ABC subgroup was noted to have an increased expression of the target genes of the transcription factor NF-kB. Davis et al. demonstrated constitutive activation of NF-kB in ABC-derived cell lines and when the NF-kB pathway was inhibited the cells underwent apoptosis. Inhibitors of the NF-kB pathway are at present being tested in clinical trials. Another possible therapeutic target could be cyclin D1 that is overexpressed in MCL patients. Inhibitors, like a p16INK4a -mimetic, could disrupt cyclin D1/CDK4 complexes should theoretically prolong the survival of MCL patients. As mentioned earlier, Elenitoba-Johnson et al. also suggested that inhibitors of p38MAPK could be used to treat transformed FL. [103] Thus, with reliable and verified GEPs defining molecularly distinct classes and subtypes of lymphomas, optimized mechanism based therapies should be an achievable future goal. Acknowledgments This work was supported in part by an NCI grant (CA84967), Department of Health and Human Services. We would like to thank members of LLMPP consortium for their contribution in field of lymphoma-genomics. We apologize to the investigators whose work was not cited due to space limitations.References
Copyright 2007 - Indian Journal of Cancer The following images related to this document are available:Photo images[cn07012f4.jpg] [cn07012f8.jpg] [cn07012f3.jpg] [cn07012f6.jpg] [cn07012f7b.jpg] [cn07012f5.jpg] [cn07012t1.jpg] [cn07012f2.jpg] [cn07012f7a.jpg] [cn07012f1.jpg] |
| |||||||||

![[Figure - 1]](/showimage?cn/photo/cn07012f1.jpg){kind=link}
![[Figure - 2]](/showimage?cn/photo/cn07012f2.jpg){kind=link}
![[Figure - 3]](/showimage?cn/photo/cn07012f3.jpg){kind=link}
![[Figure - 4]](/showimage?cn/photo/cn07012f4.jpg){kind=link}
![[Figure - 5]](/showimage?cn/photo/cn07012f5.jpg){kind=link}
![[Table - 1]](/showimage?cn/photo/cn07012t1.jpg){kind=link}
![[Figure - 6]](/showimage?cn/photo/cn07012f6.jpg){kind=link}
{kind=link}
{kind=link}