 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
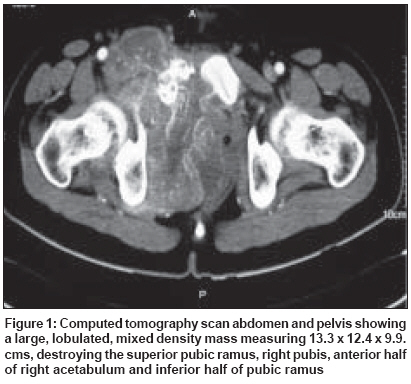
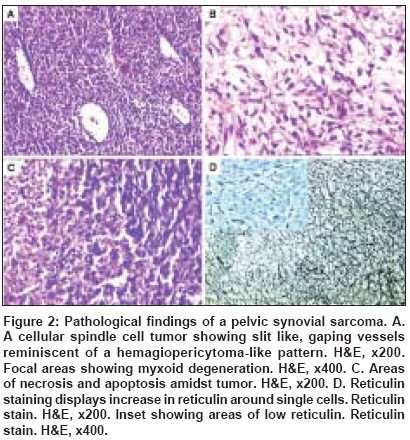
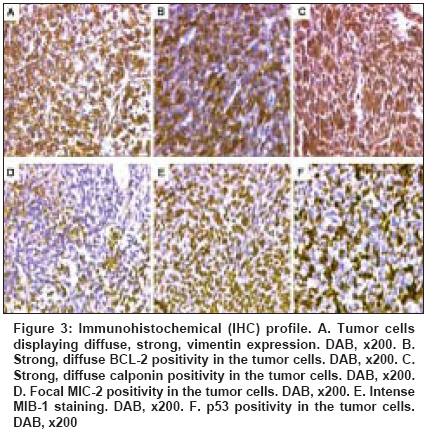
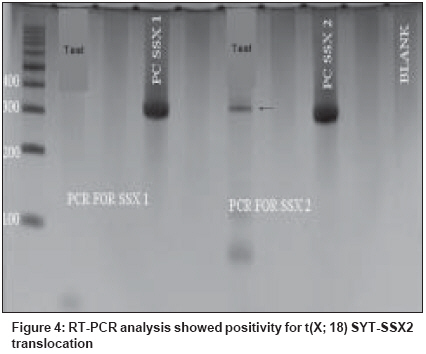
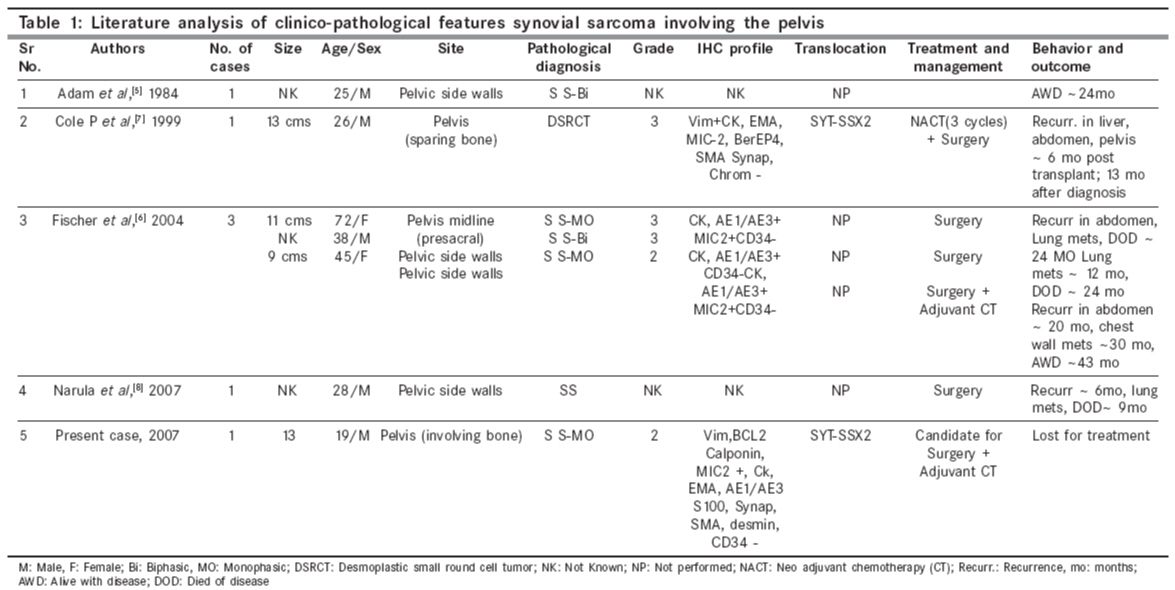
Indian Journal of Cancer, Vol. 45, No. 2, April-June, 2008, pp. 67-71 Case Report A t(X; 18) SYT-SSX2 positive synovial sarcoma in the pelvis of a young adult male: A rare case report with review of literature Rekhi B, Jambhekar NA, Desai SB, Basak R, Puri A, Agrawal M Department of Pathology, Tata Memorial Center, Mumbai Code Number: cn08018 Abstract Synovial sarcoma is uncommonly documented in the pelvis. Rarely, such cases have dealt with molecular analysis. A 19-year-old boy presented with pain and swelling in his left lower limb of two months duration. He developed acute urinary retention four days prior to his hospital admission, wherein radiological examination unraveled a large soft tissue mass, displacing his pelvic muscles, along with a lytic lesion involving his right pubic bone. Biopsy showed a cellular spindle cell sarcoma, exhibiting hemangiopericytoma-like vascular pattern with focal necrosis. Immunohistochemistry (IHC) showed positivity for vimentin, BCL-2, calponin and MIC 2. Cytokeratin (CK) and epithelial membrane antigen (EMA) were negative. MIB 1 count was 70% (high). P53 was positive. Diagnosis of a poorly differentiated synovial sarcoma was offered and confirmed with a positive t(X; 18) SYT-SSX2 translocation. This case highlights the value of molecular analysis in diagnosis of a synovial sarcoma at rare sites, especially when IHC results are equivocal and the biopsy material is limited. Keywords: Molecular analysis, pelvic synovial sarcoma, pelvic tumors, soft tissue sarcomas, t(X; 18) SYT-SSX2 translocation. Introduction Synovial sarcoma (SS) is defined as a mesenchymal spindle cell tumor, displaying variable epithelial differentiation and is characterized by a specific chromosomal translocation t(X: 18) (p11; q11). [1] It is uncommon, accounts for 5-10% of soft tissue sarcomas and is unrelated to the synovium. Traditionally, extremities form the commonest sites of its occurrence in 80-90% cases. [2] However, with the advent of ancillary techniques, it has been identified at unusual locations like head and neck region, lung, prostate. [3],[4] Few cases of SS have been documented in the pelvis, especially involving the bone. [5],[6],[7],[8] Still rare is its objective identification with molecular results in this location. [7] Herein, we report a t (X; 18) SYT-SSX2 positive, poorly differentiated synovial sarcoma in the pelvis of a young adult male, describing the value of molecular analysis in ascertaining this diagnosis.Case Report A 19-year-old boy presented with swelling and pain in his left lower limb of two months duration. Four days prior to his hospital admission, he developed acute urine retention. Subsequently, he underwent radiological evaluation. X-ray pelvis showed a permeative lesion involving his right pubic bone, superior and inferior pubic ramus, including a transverse fracture. In addition, a soft tissue mass was seen displacing the obturator and psoas fat planes. Ultra fast slice plain and contrast enhanced computed tomography (CT) scan showed a highly vascular, large, lobulated, mixed density mass measuring 13.3 x 12.4 x 9.9. cm with speck of calcification, in the right inferolateral aspect of urinary bladder, extending into the upper thigh. It was seen destroying the pubic bones and the anterior half of right acetabulum. Multiple non-enhancing areas, within the lesion, suggestive of cystic and necrotic components, were noted. Anteriorly, the lesion extended till the pubic symphisis and posteriorly, up to the anterior surface of sacrum [Figure - 1]. All the visceral organs were normal. A diagnosis of ′Ewing′ sarcoma was suggested. Subsequently, a core needle biopsy was performed and the tissue was submitted for histopathological diagnosis. Histopathological findings H and E stained sections showed a cellular tumor comprising oval to spindly cells, displaying ′stag-horn′ arrangement of vasculature, reminiscent of a hemangiopericytoma-like (HPC) pattern. Focal areas showed myxoid change, necrosis and apoptosis. Mitoses were 7/10 high power field (hpf). Reticulin stain highlighted HPC pattern and reticulin deposition around single and cell groups along with focal reticulin low areas (inset). A provisional diagnosis of a high grade, poorly differentiated synovial sarcoma of spindle cell type was formed [Figure 2 A-D] IHC showed diffuse positivity for vimentin, bcl-2, calponin and negativity for cytokeratin (CK), EMA, AE1/AE3, S100, synaptophysin, desmin, SMA, CKIT and CD34, the latter most that highlighted the vascular pattern. MIB 1 was 70% (high). P53 was positive. MIC 2 showed weak, focal positivity [Figure 3A-F] A diagnosis of a high grade, poorly differentiated, synovial sarcoma of spindle cell type was offered. Molecular analysis SYT (Forward): 5´ CCA GCA GAG GCC TTA TGG ATA 3´ The PCR products were analyzed on 10% Polyacrylamide gel, which showed a positive band for t(X; 18) SYT-SSX2 translocation. Diagnosis of synovial sarcoma was confirmed [Figure - 4]. The patient was a candidate for surgery with adjuvant CT. However, unfortunately, he was lost for treatment. Discussion Synovial sarcoma (SS) forms a distinct clinical, morphological and a genetic type of a soft tissue sarcoma, which has been described in various body sites. [3],[4],[5] Intraabdominal primary SS is unusual. Nearly 50 cases have been documented so far. Still rare, is a pelvic location, wherein only five cases have been identified to the best of our knowledge. [5],[6],[7],[8] Only one such case has dealt with molecular analysis. [7] The present case of a SS was seen in a young boy, whose radiological evaluation unraveled a large, bone-destructive pelvic mass, a feature that has not been seen with the similar documented cases. Moreover, he constitutes the youngest of all such cases, wherein the age ranged from 25-72 years [6],[7],[8] [Table - 1]. Radiologically, a pelvic SS reveals an admixture of cystic and necrotic elements, leading to a "bowl of fruit" sign, as seen in the present case. [8] This led to a putative diagnosis of a Ewing′s sarcoma. Biopsy showed a high grade, spindle cell sarcoma, which led to a range of differential diagnoses, including a malignant peripheral nerve sheath tumor (MPNST), Ewing sarcoma, a leiomyosarcoma (LMS), a gastrointestinal stromal tumor (GIST), a desmoplastic small round cell tumor (DSRCT) and a sarcomatoid mesothelioma, as noted in earlier reports. [7],[8] Despite an X-ray appearance of a permeative lesion and cytomorphological presence of oval cells, conspicuous presence of short spindly cells, made diagnosis of Ewing sarcoma, less likely. The presence of myxoid areas led to MPNST, LMS and GIST as other differentials. While lack of wavy/ ′serpentine′ nuclei was helpful in ruling out an MPNST; lack of ′cigar-shaped′ nuclei with blunt ends made diagnosis of a LMS, less likely. Negative S-100, desmin and SMA expression helped in ruling out these possibilities. GIST and DSRCT were included as other differentials, as seen earlier. [7] Lack of epithelial markers (CK, EMA and AE1/AE3), made DSRCT as a less likely possibility. Lack of epithelial marker expression in a SS was also noted by Cole et al [7] but this finding was in contrast to other studies. [6],[7] In a series of 20 cases of poorly differentiated SS, van de Rijn M [9] identified EMA positivity in 92% cases and CK in 42% cases. This explains a rare IHC profile in our case. At the same time, there is a possibility that negative epithelial marker expression might have been due to limited biopsy material in our case. Nonetheless, BCL2 and calponin positivity with focal MIC-2 expression helped to reinforce this diagnosis, as noted by others. [9],[10],[11] GIST was ruled out in view of CKIT and CD34 negativity. [10] In view of equivocal IHC results, molecular analysis was recommended, which showed t(X; 18) SYT-SSX2 positive transcript. SS has been shown to consistently express a t(X; 18; p 11; q11) translocation, which usually represents either of the 2 gene fusions, SYT-SSX1 or SYT-SSX2 that encode specific putative transcriptional proteins. [12],[13] Apart for its diagnostic value, this translocation was believed to have a prognostic value. Compared to SYT-SSX2 fusion, mostly seen in monophasic SS, a SYT-SSX1 positive transcript, more commonly seen in a biphasic SS, was associated with a relatively unfavorable prognosis. [14] However, lately studies, including the one by Guillou et al [15] have observed lesser prognostic value of specific transcript in comparison to tumor grade. Even though, the present case of a pelvic SS of spindle cell type, like the one described by Cole et al [7] exhibited similar SYT-SSX2 transcript, the other parameters forecast an unfavorable prognosis. Apart from site, larger tumor size, poor differentiation, necrosis, apoptosis along with high mitoses are indicators of a grim prognosis, as noted earlier. [1],[6],[7],[16] As per literature, it has been seen that invariably, pelvic SSs are associated with a dismal outcome as a result of local recurrences and metastasis, latter that has been noted in 4/6 similar documented cases. [6],[7],[8] Surgery is the treatment mainstay. However, marginal clearance is difficult to achieve in this location. Adjuvant chemotherapy (CT) and radiotherapy (RT) have been given. [6],[8],[16] Despite that, recurrences and metastasis have been noted. Our case is a candidate for surgery with adjuvant CT. In a nutshell, this case reinforces value of molecular analysis in a SS at uncommon sites. References
Copyright 2008 - Indian Journal of Cancer The following images related to this document are available:Photo images[cn08018f2.jpg] [cn08018f3.jpg] [cn08018f4.jpg] [cn08018t1.jpg] [cn08018f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}