 |
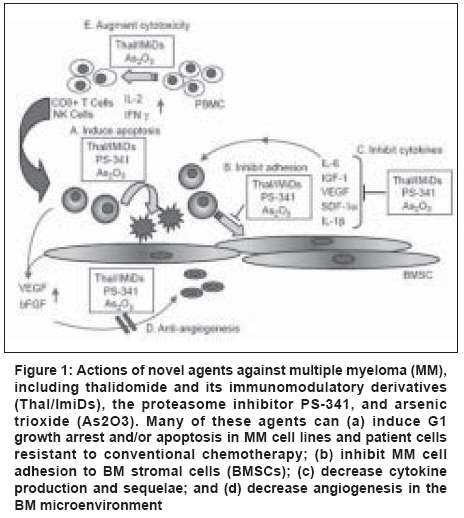
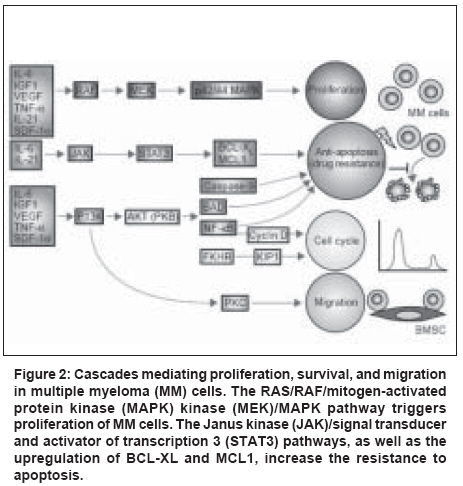
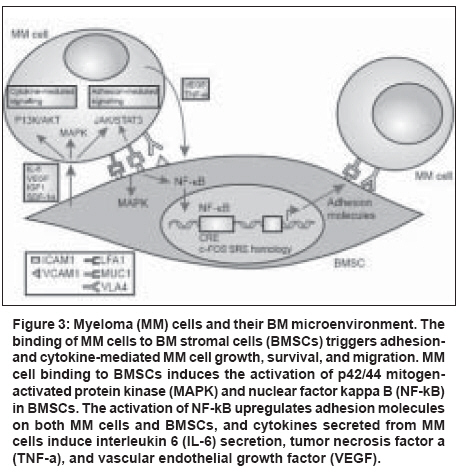
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Cancer, Vol. 45, No. 4, October-December, 2008, pp. 142-148 Review Article Newer therapeutic molecules for multiple myeloma Jain P, Gupta S, Parikh PM Department of Medical Oncology, Tata Memorial Hospital, Dr. E. Borges Road, Parel, Mumbai - 400 012 Code Number: cn08043 Abstract Therapeutic management of multiple myeloma (MM) for the last several decades has mainly involved regimens based on use of glucocorticoids and cytotoxic chemotherapeutics. Despite progress in delineating the activity of such regimens, at either conventional or high doses, MM has remained an incurable disease. This has sparked major interest in the development of novel therapies that in part capitalize on recent advances in our understanding of the biology of MM, including the molecular mechanisms by which MM cell-host bone marrow (BM) interactions regulate tumor-cell growth, survival, and drug resistance in the BM milieu. Herein, we review the latest progress in the development of these novel anti-MM therapies, with major focus on therapies which have translated from preclinical evaluation to clinical application, including thalidomide and its more potent immunomodulatory derivatives (IMiD), the first-in-class proteasome inhibitor bortezomib (formerly known as PS-341). Search strategy included Medline using the terms 'Myeloma and Newer Drugs' citations relevant to treatment guidelines issued in 1999 and 2008 were screened.Keywords: Bortezomib, multiple myeloma, thalidomide Introduction Multiple myeloma (MM) remains incurable with conventional treatments, with median survival duration of 3-5 years. The disease follows a relapsing course in the majority of patients, regardless of treatment regimen or initial response to treatment. Novel, more effective treatment approaches are required in order to improve outcomes and extend survival. [1] Newer therapies capitalize on recent advances in our understanding of the biology of MM, including the molecular mechanisms by which MM cell-host bone marrow (BM) interactions regulate tumor-cell growth, survival, and drug resistance in the BM milieu. The development of in vitro and in vivo models of MM stromal interactions has allowed not only for better characterization of these molecular phenomena but also for identification of specific therapeutic strategies to overcome these interactions and achieve an enhanced anti-MM effect, even against MM resistant to conventional therapies. Novel therapies are translating from preclinical evaluation to clinical application.Potential Targets The signaling cascades involved in the cytokine-mediated proliferation, survival, drug resistance, and migration of MM cells have been well characterized. For example, certain cytokines - e.g., interleukin-6 (IL-6) and vascular endothelial growth factor (VEGF) - trigger proliferation of MM cells via mitogen-activated protein kinase (MAPK) signaling, whereas MM cell migration induced by cytokines (e.g., VEGF) is mediated via a protein kinase C-dependent, extra cellular signal-related kinase-independent pathway. MM cell apoptosis induced by Fas, gamma-irradiation (IR), and dexamethasone is mediated by distinct signaling cascades [Figure - 1],[Figure - 2],[Figure - 3]. [2] Dexamethasone triggered (but not IR- or Fas-triggered) apoptosis is mediated via activation of related adhesion focal tyrosine kinase (RAFTK). While dexamethasone activates caspase 9, novel agents such as thalidomide and other immunomodulatory derivatives (IMiDs) deactivate caspase 8 and downstream cascades. This supports the utility of coupling novel and conventional therapies, such as thalidomide and dexamethasone. Mechanisms of drug resistance induced by cytokines have been identified: IL-6 and Insulin-like growth factor 1 (IGF-1) inhibit apoptosis triggered by dexamethasone (but not IR) via Akt signaling and nuclear factor kappa B (NF-kB) activation, with downstream induction of intracellular inhibitors of apoptosis (IAPs), including FLICE (Fas-associated death domain-like interleukin-1b-converting enzyme) inhibitor protein (FLIP), survivin, cellular inhibitor-of-apoptosis protein (cIAP)-2, A1/bfl-1, and XIAP. These studies validate signaling cascades triggered by MM localization in the BM microenvironment mediating tumor-cell growth, survival, drug resistance, and migration as potential novel therapeutic targets. Novel Agents Thalidomide and immunomodulatory derivatives Of these patients, 76 had relapse after high-dose chemotherapy. Thalidomide treatment lasted for a median of 80 days (range 2-465 days) and objective responses were observed in 32% of cases. These responses included decrease in serum or urine levels of monoclonal paraprotein by> 90% in eight patients (with complete remissions in two of them),> 75% in six patients,> 50% in seven patients, and> 25% in six patients. However, no significant changes in BM microvascular density were reported. At least one-third of the patients had mild or moderate constipation, weakness, fatigue, or somnolence. More severe adverse effects were infrequent (occurring in < 10% of patients), and hematologic toxicity was rare. Subsequent studies from other centers confirmed the activity of thalidomide in MM, [3] showing that the optimal dose of thalidomide is variable among patients and should be titrated to a level which is both well tolerated and achieves clinical response. Thalidomide has also been administered in combination with dexamethasone, cytotoxic chemotherapeutics, or other agents such as clarithromycin. These combinations generally achieve higher response rates than separate administration of either thalidomide or the drugs with which it is partnered, and in some cases clinical responses are observed in patients resistant to each of these drugs individually. Indeed, in some cases the side-effects can be potentially very serious, for example, thromboembolic events in patients receiving thalidomide combined with dexamethasone or, mainly, anthracyclines. Dose reductions of thalidomide (or other components of combination therapies), as well as careful selection and comprehensive monitoring of patients receiving these regimens, may facilitate the prevention of such events. [4] Seven phase I, II, and III studies of lenalidomide (also known as CC-5013 or CDC-5013 or revlimid) have been performed in patients with refractory or relapsed MM and have led to favorable clinical results. [5] A phase II multicenter randomized controlled open label study was conducted to select the lenalidomide regimen which, when used alone or in combination with dexamethasone, would provide the most promising evidence of efficacy in relapsed/refractory MM patients. In this trial, patients were randomized to oral lenalidomide at either 30 mg once daily or 15 mg twice daily for a total of six cycles, each comprising 3 weeks on therapy and 1 week off lenalidomide. The trial showed that the 30 mg once daily lenalidomide dose is better tolerated than the twice daily regimen, and that administration of lenalidomide in the context of a 3-weeks-on/1-week-off schedule is highly active with a response rate of approximately 35%. Importantly, the adverse event profile of lenalidomide is manageable and is hallmarked by very little neuropathy (< 5%). Incidence of thrombosis with lenalidomide therapy was analyzed. In a recent study, it was seen that there was an increasing trend when given with corticosteroids. [6] In patients in whom dexamethasone was added, improved response rates were seen in about 40% of cases. In the phase III setting, lenalidomide was combined with dexamethasone and is being compared with dexamethasone plus placebo in relapsed or refractory MM patients. This study has now completed accrual and final results are awaited. The precise molecular mechanisms mediating the anti-MM effects of thalidomide and the IMiDs have not been fully elucidated. This is mainly due to the enantiomeric interconversion of thalidomide and its spontaneous cleavage to multiple short-lived and difficult-to-characterize metabolites, as well as its species-specific metabolic activation, mainly by the liver, which confound the interpretation of preclinical in vitro and in vivo mechanistic studies. At least four distinct, but potentially complementary, mechanisms have been proposed to account, at least in part, for the antitumor activity of thalidomide and its derivatives: (a) direct antiproliferative/proapoptotic effects against MM cells, which are probably mediated by one or more in vivo metabolites of thalidomide, and include inhibition of the transcriptional activity of NF-kB and its antiapoptotic target genes - e.g., the caspase inhibitors FLIP, cIAP-2, or the antiapoptotic Bcl-2 family member A1/Bfl; [18] (b) indirect targeting of MM cells by abrogation of the tumor cell protection conferred by their cell adhesion molecule or cytokine mediated (e.g., IL-6-mediated) interactions with BM stromal cells (BMSCs); (c) antiangiogenic effects; and (d) immunomodulatory effects, including enhanced natural killer (NK) cell-mediated cytotoxicity against tumor cells. Since NF-kB protects MM cells from the proapoptotic effects of steroids or cytotoxic chemotherapeutics, the inhibition of NF-kB activity by thalidomide/derivatives may account for the ability of combinations with dexamethasone or cytotoxic chemotherapeutics to achieve more potent in vivo antitumor responses than either agent alone. [7] Proteasome Inhibitors Bortezomib (formerly PS-341) Bortezomib directly inhibits proliferation and induces apoptosis of human MM cell lines and freshly isolated patient-derived MM cells, even in MM cells resistant to conventional therapies. [8] Importantly, the in vitro anti-MM activity of bortezomib is exerted even when MM cells are exposed to the drug in the presence of BMSCs, indicating that the protection afforded to MM cells by BMSCs against cytotoxic chemotherapeutics or steroids does not apply in the setting of treatment with proteasome inhibitors. Consistent with this result, bortezomib does not confer, at the proteomic level, any global nonspecific accumulation of undegraded proteins, since the levels of many proteins remain unchanged. This can be attributed to the fact that bortezomib does not indiscriminately block all aspects of proteasome function, but instead selectively targets its chymotryptic activity. This also raises the notion that the therapeutic window associated with this class of agent may be due to the fact that, among the broad spectrum of proteolytic activities and corresponding targets of the proteasome, only a subset of them are modulated by bortezomib. A qualitative analysis of gene expression profiles of bortezomib-treated MM cells indicates that this agent causes an upregulation of proapoptotic regulators, such as the TNF-a-related apoptosis-inducing ligand (TRAIL) receptor, and suppression of antiapoptotic proteins such as survivin and Bcl-2. For many of these antiapoptotic regulators, e.g. caspase inhibitors, their decreased expression is consistent with the inhibitory effect of bortezomib on NF-kB activity; as shown with NF-kB DNA binding enzyme-linked immunosorbent assays (ELISAs), bortezomib suppresses the transcriptional activity of NF-kB due to the lack of degradation of ubiquitinated IkB. [9] The accumulation of this negative regulator of NF-kB prevents its nuclear translocation and thus suppresses the expression of NF-kB-dependent antiapoptotic genes, such as the aforementioned caspase inhibitors. Furthermore, bortezomib causes a significant reduction in the expression of transcripts for molecules implicated in cell-cycle regulation and growth-factor-induced cell signaling pathways mediating proliferation, survival, and/or drug resistance of MM cells, respectively, culminating in the induction of apoptosis via a dual activation of caspase-8 and caspase-9. In addition, bortezomib triggers significant reduction in several regulators of DNA synthesis and DNA repair. Proteasome inhibitors represent a second class of therapeutics targeting the MM cell in its BM microenvironment. Based upon studies showing that IL-6 is the major growth and survival factor for human MM cells, and the observation that MM cell adhesion to BMSCs triggers the transcription and secretion of IL-6 in BMSCs via an NF-kB-dependent mechanism, it is hypothesized that the blockade of NF-kB using bortezomib may mediate anti-MM activity by inhibiting paracrine IL-6 production in BMSCs. Studies confirm that bortezomib acts directly to induce apoptosis of MM cells resistant to known conventional therapies, overcomes the protective effects of IL-6, and adds to the anti-MM effects of dexamethasone. In the BM microenvironment, bortezomib actively inhibits the binding of MM cells to BMSCs, the transcription and secretion of IL-6 triggered by the adhesion of MM cells to BMSCs, and BM angiogenesis. These studies, coupled with phase I trials of bortezomib which demonstrated objective clinical responses in eight out of eight patients with refractory MM, provided the rationale for a multicenter phase II trial of bortezomib in MM. In that trial of bortezomib administration to 202 heavily pretreated patients with relapsed refractory MM, the overall response rate (minimal response plus partial response (PR) plus CR) was 35%, with an additional 24% experiencing stable disease. Importantly, according to Blade´ criteria, complete responses were observed in 10% of bortezomib-treated patients - 4% were immunofixation-negative and the other 6% had residual MM paraprotein levels detectable only by immunofixation. Median time to progression was seven months and median duration of survival was 16 months, with a median duration of response of 12 months in those patients with PR and CR. Paraprotein responses to bortezomib treatment were associated with improved hemoglobin levels, decreased blood transfusion requirements, improvement in renal function, and normalization of uninvolved immunoglobulins. Drug-related gastrointestinal toxicity and fatigue were manageable in most cases. Significant thrombocytopenia and neuropathy occurred primarily in patients in whom these conditions were pre-existent, and serious adverse events were uncommon. Importantly, recent data from a multicenter, international phase III trial in relapsed MM patients - which included 669 patients from 93 centers - indicate that bortezomib is superior to high-dose dexamethasone therapy, in terms of both time to disease progression and overall survival, as well as response rate (45 versus 26%, P < 0.0001). [10] Importantly, superiority for bortezomib versus high-dose dexamethasone was observed in second line (i.e. at first relapse) and as later salvage therapy, with toxicity profiles between the two treatment arms that were balanced and manageable. In combination with pegylated liposomal doxorubicin it significantly improved TTP compared with bortezomib alone in relapsed refractory MM. [11] Second generation proteasome inhibitors are under phase II trial, while PR-171, a novel epoxyketone-based irreversible proteasome inhibitor, is currently in clinical development. In comparison to bortezomib, PR-171 exhibits equal potency but greater selectivity for the chymotrypsin-like activity of the proteasome. PR-171 is well tolerated when administered for either 2 or 5 consecutive days at doses resulting in> 80% proteasome inhibition in blood and most tissues. In human tumor xenograft models, PR-171 mediates an antitumor response that is both dose and schedule dependent. The antitumor efficacy of PR-171 delivered on 2 consecutive days is stronger than that of bortezomib administered on its clinical dosing schedule. [12] VEGF inhibitors TRAIL/Apo2L Farnesyl transferase inhibitors Recently, the activity and tolerability of the FTase inhibitor tipifarnib (Zarnestra) was evaluated in a phase II clinical trial in patients with relapsed MM. Forty three patients who had previously received a median of four (range 1-6) chemotherapy regimens received tipifarnib at 300 mg orally twice daily for 3 weeks every 4 weeks. The most common toxicity was fatigue, occurring in 66 %of patients. Other adverse effects included diarrhea, nausea, neuropathy, anemia, and thrombocytopenia; 64% of patients had disease stabilization. Treatment with tipifarnib suppressed FTase, but not GGTase-I, in BM and peripheral-blood mononuclear cells. However, inhibition of farnesylation did not correlate with disease stabilization. These data suggested that FTase inhibitors can be tolerable and induce disease stabilization in relapsed MM patients. [15] Additionally, it was demonstrated that combination of a bortezomib and lonafarnib demonstrates synergistic myeloma-cell death and warrants further preclinical and clinical studies. [16] Histone deacetylase inhibitors The therapeutic targeting of HDAC function has been translated into clinical trials with the use of hydroxamic-acid-based hybrid polar compounds, such as Vorinostat (suberoylanilide hydroxamic acid (SAHA)). SAHA induces accumulation of acetylated core nucleosomal histones, with related induction of differentiation and/or apoptosis in transformed and neoplastic cells. SAHA treatment of MM cells was associated with increased p21 and p53 protein levels, dephosphorylation of Rb, cleavage of Bid (which suggests a role for Bcl-2 family members in regulation of SAHA induced cell death), as well as a calpain-dependent, caspase-independent pathway for induction of apoptosis in MM cells. Importantly, SAHA sensitizes MM cells to a wide range of proapoptotic stimuli, including death receptor ligands such as FasL and TRAIL/Apo2L, 79dexamethasone, cytotoxic chemotherapeutics, thalidomide analogs, and bortezomib. [17],[18] Multifaceted anti-MM effects can be attributed to the constellation of antiproliferative and/or proapoptotic molecular events triggered by SAHA, including downregulation of transcripts for members of the IGF/IGF-1 receptor (IGF-1R) and IL-6 receptor (IL-6R) signaling cascades, antiapoptotic molecules (e.g., caspase inhibitors), oncogenic kinases, DNA synthesis/repair enzymes, and transcription factors (e.g., XBP-1, E2F-1) implicated in MM pathophysiology, as well as suppression of activity of the proteasome and expression of its subunits. Heat shock protein 90 inhibitors Hsp90 inhibition also blocks both constitutive and cytokine-induced activation of NF-kB and telomerase in the BM milieu. As a result of these properties, GA and other Hsp90 inhibitors induce apoptosis of MM cell lines and primary MM cells which are resistant to dexamethasone, anthracyclines, thalidomide, IMiDs, TRAIL/Apo2L, and bortezomib. Interestingly, bortezomib treatment of MM cells induces a major compensatory upregulation of Hsp90, which represents a stress response in tumor cells to withstand the toxic effects of intracellular accumulation of undegraded proteins. Therefore, blocking this response with hsp90 inhibitor (Tanespimycin) enhances MM cell apoptosis triggered by bortezomib, suggesting a possible role of combination regimens with these two classes of drugs in MM. [19] References
Copyright 2008 - Indian Journal of Cancer The following images related to this document are available:Photo images[cn08043f3.jpg] [cn08043f2.jpg] [cn08043f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}