 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
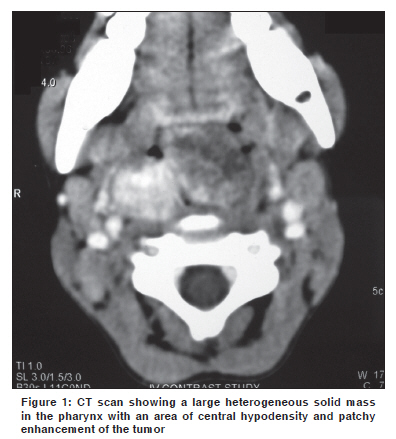
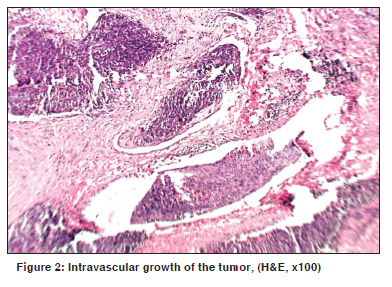
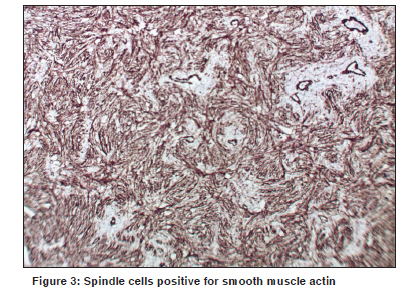
Indian Journal of Cancer, Vol. 47, No. 1, January-March, 2010, pp. 78-79 Letter To Editor Infantile myofibroma of the pharynx presenting with severe upper airway obstruction in a child Panja S, Champaka G1, Shenoy AM Department of Head and Neck Oncology, Department of Pathology, Kidwai Memorial Institute of Oncology, India. Code Number: cn10021 PMID: 20071800 DOI: 10.4103/0019-509X.58869 Sir, A six-year-old girl presented to us with a history of difficulty in swallowing and occasional blood tinged sputum for two months. On examination a pinkish, lobulated mass, with ulceration, was noticed in the oropharynx extending down to the hypopharynx, occluding the airway. At presentation she had severe respiratory distress and stridor for which an emergency tracheostomy was done. Computerized tomography showed a large heterogeneous solid mass in the retropharyngeal area with central hypodensity and patchy enhancement of the tumor, more on the right side [Figure - 1]. The child was planned for biopsy under general anesthesia, to establish the diagnosis. Intraoperatively a lobulated mass was seen rising from the posterior pharyngeal wall; attached to the right side, and extending to the hypopharynx. The tumor was excised completely. The specimen consisted of a nodular mass measuring 4 x 3 x 1 cm. The cut section was grayish-white and homogenous, with areas of hemorrhage. Microscopy revealed a neoplasm with multinodular proliferation of oval to spindle cells arranged in whorls around the blood vessels. The oval cells had vesicular nuclei with one or two small nucleoli and a moderate cytoplasm. Mild-to-moderate atypia was noted. The spindle cells had elongated tapering nuclei and scanty cytoplasm. The mitotic count was 6 to 7 / 10 hpfs in the mitotically active areas. Intravascular growth of the tumor was noted [Figure - 2]. The stroma also showed an infiltrate of lymphocytes and plasma cells. On immunohistochemistry, the tumor cells were positive for smooth muscle actin [Figure - 3] and negative for S-100, CK, EMA, CD23, CD34, Desmin, MyoD1, HMB-45, and LCA. The child was successfully decannulated postoperatively and has been kept under regular follow-up. There has been no evidence of recurrence over the last ten months of follow-up. Infantile myofibromatosis of head and neck are extremely rare, especially in the Indian population. First described in 1954, by Stout, [1] the term "Infantile myofibromatosis" was coined by Chung and Enzinger as a distinct lesion, based on its unique clinical and staining characteristics consistent with its myofibroblastic origin. [2] Although the etiology is unclear, it is believed to be genetic in nature. Microscopically, they form well-circumscribed nodules of plump spindle cells, displaying characteristics intermediate between fibroblasts and smooth muscle cells. Immunophenotypically, the myofibroblastic component and the more primitive component are positive for SMA. Behavior-wise, some myofibromas regress spontaneously. Chung and Enzinger reported a 10% recurrence rate for the lesions they reviewed, but there do not appear to be any factors that are predictive of a recurrence. [2] However, the multicentric variant has a much poorer prognosis than the solitary one. [3] In cases where the tumors are life threatening, treatment involves surgical excision or low-dose chemotherapy as well as palliative care for symptoms. On account of rapid proliferation and features of increased cellularity and extensive necrosis, these tumors can easily be misdiagnosed as malignant neoplasms. [4] Large lesions located in the aerodigestive tract can unusually present with severe airway compromise in children. [5] The rarity of the disease and the possibility of missing the diagnosis of clinically mild cases makes it difficult to identify and treat. References
Copyright 2010 - Indian Journal of Cancer The following images related to this document are available:Photo images[cn10021f1.jpg] [cn10021f3.jpg] [cn10021f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}