 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Cancer Research and Therapeutics, Vol. 1, No. 3, July-September, 2005, pp. 151-161 Original Article Heterogeneity in the radiosensitizing effects of the DNA ligand hoechst-33342 in human tumor cell lines. Adhikari JS1, Khaitan Divya1, Arya MB2, Dwarakanath BS1 1Division of Biocybernetics, Institute of Nuclear Medicine and Allied Sciences, Delhi, India and 2PG DAV College, Hapur, India
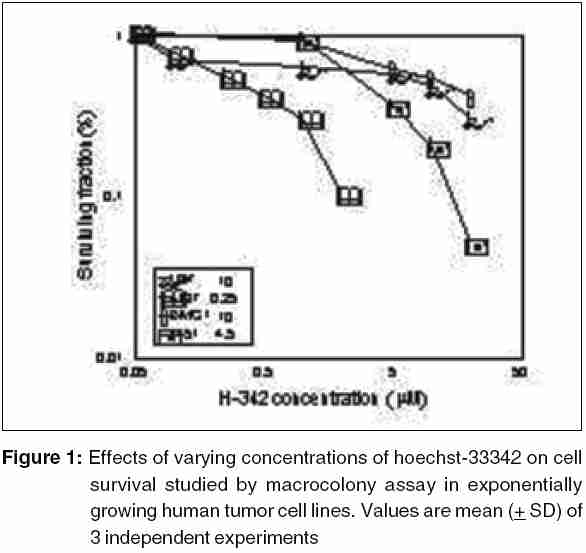
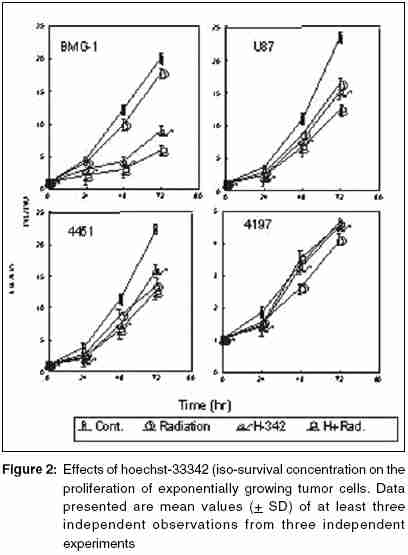
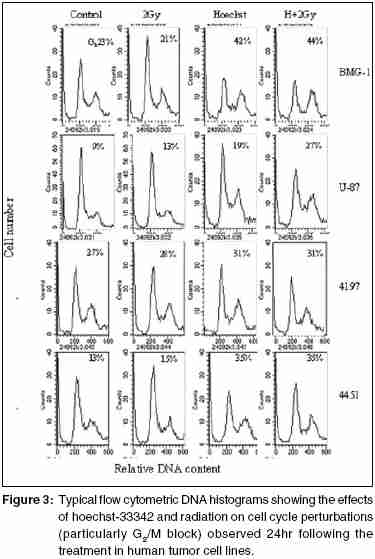
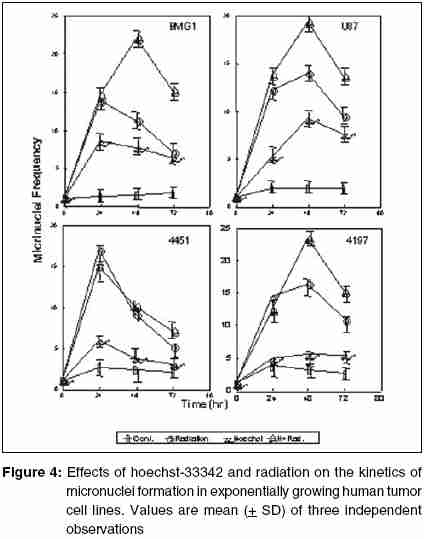
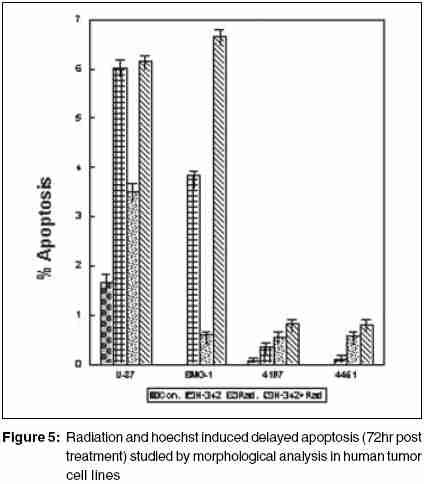
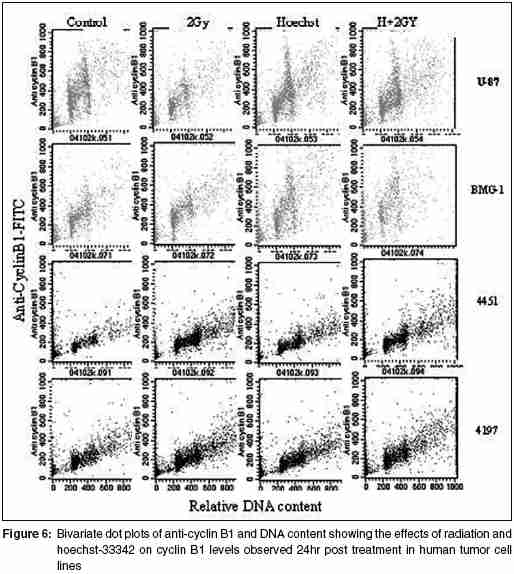
Code Number: cr05035 Abstract The AT specific minor grove DNA binding ligands bisbenzimidazole derivatives like hoechst-33342 and hoechst-33258 which scavenge free radicals and stabilize macromolecular structure have been shown to afford radioprotection by reducing the induction of DNA damage. However, their ability to inhibit topoisomerases I & II, which play important roles in damage response pathways including DNA repair can enhance radiation damage under certain conditions. Since pool sizes of the topoisomerases differ not only between normal and tumor cells, but also among different tumors, it is anticipated that radiosensitization by hoechst-33342 can vary among tumors. The present studies were, therefore, undertaken to verify this proposition in human glioma (BMG-1 & U-87) and squamous carcinoma (4197 & 4451) cell lines which differ in their biological behavior (ploidy, p53, cyclins, bcl, bax etc).Isotoxic concentrations of hoechst-33342 (IC50 i.e producing 50% cell kill) administered immediately following irradiation resulted in the radiosensitization of all cell lines, with a 4&7 fold increase in the cell death (loss of clonogenic cell survival) in U-87& BMG-1 and a 3 fold increase in 4197 &4451 cells. Growth inhibition and increase in cytogenetic damage (micronuclei formation) as well as delayed apoptosis observed under these conditions corroborated well with the enhanced cell death. The ligand induced a significant cell cycle delay, particularly in the late S and G2 phases of BMG-1, U-87 and 4197 cells, while no significant changes could be observed in 4451 cells. Higher endogenous levels of cyclin B1 found in both the glioma cell lines, was enhanced further by the ligand as compared to the squamous carcinoma cells. These results clearly demonstrate that the radiosensitizing effects of the ligand are indeed heterogeneous among different human tumor cell lines. The radiaosensitization is p53 independent and accompanied by enhanced mitotic death (linked to cytogenetic damage) as well as induction of cyclin B1 mediated apoptosis.Keywords: Radiosensitization, Topoisomerase inhibitors, DNA ligand, Hoechst-33342, Apoptosi, CyclinB1 INTRODUCTION Radiation therapy either alone or in combination with surgery is employed in treating majority of the human tumors. However, its success is limited by inherent resistance of certain tumors, presence hypoxic cells and heterogeneous population with varying degrees of radio-sensitivity, besides limited tolerance of normal tissues. Therefore, development of approaches and agents, which selectively sensitize tumors continue to be a major focus of research in experimental radiation oncology. Towards this end, combined modality employing different variety of chemotherapeutic agents and ionizing radiation is one of the widely used strategy with enhanced therapeutic efficacy, particularly in the form of local tumor control as compared to either of the modality when used alone.[1],[2] In addition to their inherent cytotoxicity, certain chemotherapeutic drugs have been found to enhance radiation-induced cell death i.e radiosensitization,[3],[4] which could be partly responsible for the improved tumor regression. Since the mechanisms of cytotoxicity differ among these agents, although most act through the induction of DNA lesions, an insight in to the interactions between these chemotherapeutic drugs and radiation is expected to greatly facilitate the development of more potent radiosensitizers for improving cancer therapy.The radiation response of mammalian cell is characterized by induction and repair of DNA damage, cell cycle progression, alteration in signal transduction and apoptosis. Since DNA damage, influences of all these processes, induction and repair of DNA damage are considered the most important determinants of radiation response influencing cell death, mutation and transformation.[5],[6],[7] Both these processes are influenced by a number of physico-chemical and biological factors that include the nature and dose of radiation, oxygen tension, levels of free radical scavengers, pool sizes of the repair enzyme and nucleotides, cell cycle position and the status of chromatin organization,[8],[9],[10],[11] While damage induction by low LET radiation is significantly higher in DNA associated with relatively more open chromatin structure,[12] several events in the repair like damage recognition and recruitment of repair elements are greatly influenced by the chromatin status. Although acetylation of histones has been shown to profoundly influence the degree of chromatin condensation, topological modifications of DNA, regulated by topoisomerases also play an important role in maintaining the optimal status during several DNA transactions like replication, transcription and repair.[13] Topoisomerases are a class of nuclear enzymes involved in DNA winding and unwinding thereby facilitating the progression of replication fork,[14],[15] transcriptional bubble and separation of daughter strands (chromosome segregation) during mitosis.[16] Topoisomerase II (TOP II) is an integral component of nuclear matrix and an important regulator of DNA synthesis,[17] chromosome condensation and separation.[18] On the other hand, topoisomerase I (TOP I) is believed to facilitate chromatin assembly, recombination, cell division, template reading by RNA polymerase.[14],[18] The enzymes (TOP I and TOP II) first bind to DNA in a non-covalent manner, followed by nicking of one or two strands during which the enzyme and DNA become covalently linked.[18] This hybrid an intermediate in the topological manipulation of DNA by the enzyme is called cleavable complex.[19] After rewinding of the helix, the strands are re-ligated. Many compounds that specifically stabilize the cleavable complex by interfering in the religation process have shown strong anti-proliferative and apoptotic effects and thereby currently employed as anticancer drugs.[20],[21],[22] The bisbenzimidazole derivatives hoechst (hoechst-33258 and hoechst-33342) are AT rich, minor grove-binding ligands that protect DNA against low LET ionizing radiation damage primarily due to its ability to scavenge free radicals and quench DNA radicals,[23],[24],[25] although a stabilization of macro molecular structure and displacement of DNA bound water molecules have also been suggested.[26],[27] However, the DNA bound ligand has also been shown to alter the chromatin condensation [28] and therefore interfere in the repair of DNA damage.[29],[30] Furthermore, the ligand has been shown to inhibit the functioning topoisomerases stabilizing the cleavable complexes, thereby leading to cell death.[18] Induction of protein - DNA cross-links and DNA strands breaks, G2 - phase arrest and chromosome endoreduplication are some of the prominent effects of hoechst-33342 treatment,[31] cellular effects are reminiscent of those induced by topoisomerase poisons and other DNA damaging agents.[32] Since tumor cells show elevated levels of topoisomerases, the ligand has been suggested to be a potential anticancer drug.[33] Studies in murine and human tumor cell lines have indeed shown that the ligand enhances the ionizing radiation and UV induced cytotoxicity by inhibiting DNA repair, leading to enhanced cytogenetic damage and apoptosis.[30],[34],[35],[36] In vivo studies with focally irradiated Ehrlich ascites tumors in mice have clearly shown a dose dependent radiosensitization by the ligand resulting in local tumor control and cure rates of nearly 50%.[37] Since tumor cells differ in their topoisomerase pool size as well as in the degree of chromatin condensation, the radiomodifying effects of hoechst can vary among different tumors. Therefore, in the present studies we investigated the radiomodifying effects of hoecsht-33342 in human tumor cell lines of different origin and biological behaviour. Our results in two glioma (BMG-1, U-87) and two squamous carcinoma (4451, 4197) cell lines show that the radiosensitizing effects of hoechst-33342 are heterogeneous and independent of p53 status. MATERIALS AND METHODS Cell Line The cerebral glioma cell line (BMG-1; diploid,wild type p53) was established in Bangalore, India.[38] U-87 (malignant glioma, diploid, wild type p53) was obtained from Dr. Stuschke of Radiotherapy Dept., Universitatsklinikum, Essen. Two squamous cell carcinomas (4451, hyper diploid, mutated p53 & 4197, diploid, wild type p53) established at Institute of Medical Radiation Biology Essen, Germany was kindly provided by Prof. C. Straffer of Inst,[39] of Med. Rad. Bio. Essen. Cells were maintained on Dulbecco′s Modified Eagles Medium (DMEM) supplemented with 5% fetal bovine serum for BMG-1, 10% for 4451 and U-87 and 20% for 4197, HEPES buffer (10 mM) and antibiotics (Penicillin G 50000 unit/l. Streptomycin 38850 unit/l and Nystatin 9078 unit/l). Stock culture were maintained in the exponential growth phase by passaging them every 3 days with their respective growth medium in 25 Cm2 plastic flask (Tarson, Calcutta, India). Chemicals Hoechst 33342, Dulbecco,s Minimum Essential Medium (DMEM) and all antibiotics, citric acid and tween-20, the fetal calf serum were procured from Sigma Chemical Co. (St Louis, USA). The immuno-flowcytometric reagents, primary antibody (anti cyclin B1, anti-p53) were procured from Becton Dickinson (San Jose, USA) and secondary antibody (IgG1 whole molecule with FITC conjugate) was also procured from Sigma. Phosphate buffered saline (PBS) and Hanks Balanced Salt Solution (HBSS) was obtained from Hi-Media (Mumbai, India). Experimental Procedure and Irradiation Before treatment, the growth medium was replaced with HBSS containing 5mM glucose, which does not support growth. Hoechst was added 1h prior to irradiation in the dark, and irradiated at room temperature with a co-60 source (ELDORADO-78, USA) teletherapy unit (dose rate = 1.2 Gy/min). Following treatments, hoechst was removed and cells were washed with HBSS. Respective growth media were supplemented to each flask and cells were grown for various time intervals to study the radiobiological parameters like micronuclei formation, apoptosis, clonogenic survival etc. Clonogenic Cell Survival Assay 150-1500 Cells were plated (depending on the treatment) in 60-mm petri dishes 12-15 hr prior to treatment, to facilitate the attachment of cells. After various treatments, petri dishes were incubated at 37οC in a humidified 5% CO2 atmosphere for 8-10 days. Colonies were fixed in methanol and stained with 1-% crystal violet (made in 70% methanol). Colonies containing more than 50 cells were counted. Differences between the mean values of data from different groups were tested for statistical significance by student′s t test. The modifying factor r hoechst was calculated as shown below: ρSF hoechst = [(SF) hoechst+γ]/[(SF)γ] Cell Proliferation Exponentially growing cells were plated at a density of 6000 to 8000 cell/cm2. 20-24h later growth media replaced and HBSS was added to each dish containing hoechst at iso-survival concentration for 1hr. Following irradiation the HBSS was immediately replaced, and respective growth media were added. The cells were harvested every 24 h, using 0.05% trypsin, counted using hemocytometer, and fixed in 70%-chilled ethanol for the analysis of cell cycle distribution by flow cytometry. Cell proliferation was calculated by: Radiation-induced Division Delay Ethanol-fixed cells were washed twice with PBS (Ca2+ and Mg2+ free) and treat with RNAse (200 mg/ml final conc) for 30min at 37οC. Wash the cell again with PBS and stained with propidium Iodide (final conc. of 50 um/ml). Measurements were made by Laser based Flow-cytometer (FACS Callibur, Becton Dickenson, San Jose, USA) using cell quest soft ware in list mode. Cell cycle analysis was carried out by using MOD FIT LT-2 software variety (SanJose, USA). Micronuclei Analysis Air-dried slides of acetic methanol fixed cell were stained with DNA specific fluorochrome, diamidino-2-phenylindole dihydrochloride, DAPI.[30] Approximately 3000 cells were analyzed from duplicate slides. Since radiation as well as hoechst are known to alter the rate of cell proliferation which influences the expression of micronuclei, cell numbers as well as percentage of cells with micronuclei were determined as a function of time up to 2-3 population doubling after irradiation. Data were analyzed by obtaining integrated values of micronuclei frequency and normalizing the values with respect to cell numbers as describe earlier.[38] The frequency of cells with micronuclei called the M-fraction (MF) was calculated as: MF (%) = Nm/Nt x 100, where Nm is the number of cells with micronuclei and Nt is the total number of cells analyzed. A modifying factor ρMFHoechst was calculated as shown below, to evaluate the radiomodifying effects by taking in to account the effects on cell proliferation: ρMFHoechst = [(MF) hoechst+γ-(MF)c]/[(MF)γ-(MF)c] A ρ value> 1 means an increase in the radiation damage due to the modifier, while ρ < 1 means a decrease in the damage. Apoptosis Morphologically, marked condensation and margination of chromatin, fragmentation of nuclei and cell shrinkage characterize apoptotic cells and a good correlation between these morphological changes and DNA ladder (one of the hallmarks of cells undergoing apoptosis) has been demonstrated [41]. Percentage of cells undergoing apoptosis was determined microscopically using DAPI as a DNA specific flurochrome for cells staining. At least 1000 cells were counted and percent apoptotic cells determined from slides prepared for micronuclei formation. The modifying factor ρApoHoechst was calculated similar to micronuclei to evaluate the radiomodifying effects of hoechst. Cyclin B1 Immunostaining was performed as described earlier [40]. Briefly, Ethanol fixed cells was first permeabilized with 0.25% Triton-X 100 in PBS for 5 min on ice. Cells were then washed with PBS and incubated with respective primary antibodies diluted (1:400) in PBS containing 1% BSA, (Pharmingen, a Becton Dickinson Co.) for overnight at 4°C. Cells were then washed with PBS containing 1% BSA and incubated with FITC labeled goat anti-mouse IgG1 secondary antibody (Sigma chemicals) diluted 1:100 in PBS containing 1% BSA for 30 min at 4°C. After washing, cells were treated with 0.1% RNase and propidium iodide (PI, 10mg/ml) for 30 min. Green (FITC) and red (PI) fluorescence were recorded for at least 10,000 events as described earlier. Cell cycle was also analyzed from the measurement of DNA content from PI fluorescence. RESULTS Cell survival The clonogenic potential (plating efficiency) was nearly 70% in all the cell lines investigated. hoechst-33342 is known to be cytotoxic due to its inhibitory effects on topoisomerases I and II [42],[43], the vital nuclear enzymes, whose levels could differ among different cell lines. Therefore, we first investigated the cytotoxicity of hoechst in these four cell lines. The dose response for hoechst-33342 clearly showed that 4197 cells were highly sensitive among the four cell lines investigated, with an IC50 (concentration producing 50% cell death) value of 0.25 mM followed by 4451 (IC50 = 4.5 μM), where as both the glioma cell lines were relatively less sensitive with an IC50 value of 10 mM [Figure - 1][Table - 1]. These variations in the sensitivity correlated reasonably well with the TOP I levels analyzed by the plasmid relaxation assay (Shailja et al., unpublished observations). Addition of hoechst 1 h before irradiation enhanced the radiation dose response of BMG-1 cells and nearly abolished the shoulder (data not shown) as reported earlier in human cells.[44],[45],[46] The degree of radiosensitization was dependent both on the concentration of hoechst and absorbed radiation dose. Further studies were carried out at therapeutically relevant radiation dose of 2 Gy and at the respective IC50 values for the cell lines. The ligand enhanced the radiosensitivity of all the four cell lines investigated by 3-7 folds [Table - 1], with both the glioma cell line showing a higher degree of sensitization (4-7 folds) as compared to the squamous carcinoma cell lines (~3 folds). The combined effects of hoechst and radiation was clearly synergistic in all the cell lines, as the observed SF values were significantly higher than the values expected from the additive effects [Table - 1]. Cell proliferation Since DNA damage induce cell cycle checkpoints resulting in G2block as well as G1-S transition delay, and both ionizing radiation and hoechst are known to inhibit cell proliferation we examined the growth inhibitory effects and cell cycle distribution at various time intervals after the treatments. An absorbed dose of 2Gy induced a marginal delay in the growth followed by recovery in all the cells, although the extent of delay was somewhat different among these cell lines [Figure - 2]. While hoechst had a profound effect on the growth of unirradiated as well as irradiated BMG-1 cells, a moderate effects was seen in U87 and 4451 cells, with minimal effect in 4197 [Figure - 2]. Flowcytometric analysis of the cell cycle distribution carried out at 24 h after treatment showed that radiation as well as hoechst independently induced G2 block in all the cell lines, albeit to different extents [Figure - 3]. However, the combination did not result in a higher level of G2 block in these cell lines, except in BMG-1. Measurements made at subsequent intervals up to 72 h post-treatment, suggested that under these conditions (IR dose and hoechst concentrations) the G2 block was essentially transient, as the cell cycle distributions returned to the levels in untreated cells (data not shown). Cytogenetic Damage Mitotic death (linked to cytogenetic damage) and the interphase death (mainly in the form of apoptosis) are two important modes of cell death following irradiation. Therefore, we investigated the contribution of mitotic death in the radiosensitization by analyzing the micronuclei induction in all the four cell lines. To compare the induction of micronuclei formation, the percentage of cells with micronuclei (M-fraction), when the cells have completed one post irradiation doubling (between 24-36 h post-irradiation) as well as over all induction until 72 h were analyzed. The frequency of un-irradiated cells with micronuclei (M-fraction) was in the range of 1-4%. The kinetics of micronuclei induction following an absorbed dose of 2 Gy showed a maximum frequency in the range of 10-15% among these cell lines, which peaked between 20 and 48 h post-irradiation [Figure - 4]. This was followed by a decline in the MF values, due to the well-known dilution effects.[37] Under these conditions, exposure of cells to hoechst for 1 h at IC50 concentrations induced a maximum micronuclei frequency in the range of 5-9% in these cells [Figure - 4]. Under the present experimental conditions, hoechst (1 h at IC50 concentrations) significantly enhanced the radiation induced micronuclei formation in U-87, BMG-1 and 4197 cells [Figure - 4]. The pMN values calculated at 48h post treatment were 1.54, 2.10 and 1.53 respectively. However, no significant increase was observed in 4451 cells. These results suggest that increase in the cytogenetic damage is a significant contributing factor for the enhanced radiation induced cell death by hoechst-33342 in three of the four cell lines investigated (U-87, BMG-1 and 4197). Apoptosis Induction of apoptosis was studied by the morphological analysis of cells stained with a DNA specific fluorescent dye (DAPI)) as described earlier.[30] Under the present experimental conditions a significant induction of apoptosis was observed only after 72h following irradiation (delayed apoptosis) similar to earlier observations.[30] While an enhancement in the radiation induced apoptosis was observed in hoechst-33342 treated BMG-1 cells no significant changes were observed in U-87, 4451 and 4197 cell lines. However, in U87 cells a significant induction was evident with hoechst alone 6%; [Figure - 5] under these conditions (1 h at IC50 concentrations), while the increase of radiation-induced apoptosis was insignificant. Interestingly, a significant induction of apoptosis could not be observed either with hoechst or gamma rays (2 Gy) alone or in combination in both the squamous carcinoma cell lines viz. 4451 and 4197 [Figure - 5]. Cyclin B1 Varying fractions of DNA damage carrying cells arrested in G 2as well as G1 phases have often been shown to undergo apoptosis. The induction of apoptosis particularly in G2+M cells has been shown to be cyclin B1 mediated and p53 status dependent.[47] Results presented here [Figure - 3] did show induction of G2 block as well as delayed apoptosis in some of the cell lines. Therefore, we examined the levels of endogenous as well as treatment-induced levels of cyclin B1 and p53 in these cells. Interestingly enough, both the glioma cell lines showed relatively higher endogenous levels of cyclin B1 as compared to the two squamous carcinoma cell lines [Figure - 6]. In addition, treatment induced cyclin B1 levels were also clearly elevated in both the glioma cell lines, while significant changes were not noted in the two squamous carcinoma cells. In this respect, hoechst-33342 appeared to induce a relatively higher level of cyclin B1 as compared to radiation, with no further significant increase by the combination [Figure - 6]. Therefore, it appeared that cyclin B1 could be partly responsible for the induction of apoptosis observed in the two-glioma cells. However, this induction of apoptosis did not seem to be strongly correlated with the p53 status as BMG-1, U-87 and 4197 cells carry a wild type p53 gene, while 4451 cells are mutated.[39],[48] We also examined the p53 levels following the treatment and found no significant induction in any of the cell lines (data not shown). DISCUSSION Ionizing radiation elicits a number of cellular and molecular responses such as induction and repair of DNA damage, cell cycle disturbances, programmed cell death, alterations in gene expression and signal transduction pathways.[8],[11],[49],[50],[51] Induction and repair of DNA damage are central among the several molecular targets for modifying cellular radiation responses and most often correlated well with cell death.[52],[53] DNA topoisomerases, which break and rejoin DNA in a concerted fashion thereby reliving the tension, have been recognized to play a major role in many DNA transactions, including DNA repair.[54] Several classes of compounds, which promote chromatin condensation in interphase cells, are known to modify the radiosensitivity.[55] Radiosensitization has been observed in many instances by modifiers of phosphatases, histone deacetylases, and totoisomerase inhibitors.[56],[57],[58] Although the radiosensitizing effects of the ligand has been presented here for the therapeutically relevant dose of 2 Gy, preliminary studies had shown that the ligand profoundly reduces the "shoulder" of the survival curve, while altering minimally the cell death at higher doses (> 5 Gy). These observations are similar to the results reported earlier with this ligand and camptothecin, a widely known TOP1 poison[45] in human cells.[33] TOP 1 has been reported to be the primary target responsible for the cytotoxicity induced by DNA minor groove-binding drugs including hoechst-33342.[33],[59] The magnitudes of "shoulder" in the radiation dose response curve for cell survival is a measure of the repair capacity for "sublethal damage"[60],[61] and therefore, the sensitization by the ligand could be partly due to the inhibition of the repair of sub-lethal damage. Alternatively, the ligand may saturate cellular repair processes responsible for the repair of sublethal DNA damage caused by ionizing radiation.[46] The ligand is known to stabilize the TOP1-DNA cleavable complexes in actively DNA sentisizing cells,[62] resulting in the formation of DNA damage as has been found in case of the TOP1 poison camptothecin.[62],[63] However, the radiosensitization by Hoechst-33342 differed somewhat from other TOP 1 inhibitors such as camptothecin with reference to the schedule dependency. While addition of camptothecin immediately following irradiation has been shown not to sensitize Chinese hamster DC3F cells,[45] significant sensitization has been reported with hoechst-33342 in EAT and BMG-1 cells under these conditions.[30] The precise molecular mechanism of radiosensitization of DNA topoisomerase I-targeting drugs still remains to be defined. It is possible that radiosensitization is brought about by the interaction of cleavable complexes stabilized by topoisomerase I inhibitors and a variety of primary DNA lesions induced by radiation. It has been suggested that the interaction of "potentially sublethal" cleavable complexes with cellular processes such as DNA replication, RNA transcription, and DNA repair may transform them into "sublethal" DNA damage, which upon interaction with radiation-induced DNA damage gets converted into "lethal" DNA damage.[33],[64] In this perspective, it is pertinent to note that the ligand has been shown to alter the degree of chromatin condensation[28] and inhibit transcription and replication in a concentration dependent manner.[65] These observations suggest that the bound ligand can hinder the repair of radiation-induced DNA damage and also alter the expression of genes related to the damage response pathways resulting in enhanced cell death.[66],[67] Lack of a strong correlation between the cytotoxic effects of the ligand (characterized by the IC50 values) and its radiosensitization ρ vales in [Table - 1] among the four cell lines investigated here suggests that TOP1 inhibition is not solely responsible for the radiosensitizing effects. For example, the IC50 value of U-87 cells (10 mM) was 40 times higher than that of 4197 cells (0.25 mM), while their r values were nearly identical (3.50 & 3.85). Similarly, BMG-1 and U-87 cells with identical values of IC50 differed in their rvalues by two folds [Table - 2]. Loss of clonogenic survival generally reflects anti-proliferative effects, including induction of apoptotic and nonapoptotic forms of cell death. The apparent quantitative differences in the effects of Hoechst on radiation-induced growth inhibition [Figure - 2] and loss of clonogenicity [Table - 1] could be related to the cell density dependent differences in the cellular responses, as the proliferation studies employed a higher cell density (~ 12,000 - 15,000 cells per cm2) as compared to a density of (12 - 96 cells per cm2). Mitotic death is primarily responsible for the loss of clonogenicity in most of the epithelial tumors, often leading to delayed apoptosis as a secondary event.[68] On the other hand, apoptosis contributes significantly to the radiosensitivity of hematopoietic cells, where radiation induces both intrinsic as well as extrinsic pathways of cell death.[69] Induction of apoptosis has been considered as a potential mechanism for radiosensitization, although its role in therapeutic gain has been questioned recently.[70] The present results suggest that the radiosensitization by hoechst-33342 was partly due to an increase in the cytogenetic damage linked mitotic death [Figure - 4][Table - 1], with a small increase in delayed apoptosis only in the two-glioma cell lines. Differences in the enhancement of cell death (ρSF) and micronuclei formation (ρMF) suggest that other factors such as enhancement of genomic instability may also contribute to the radiosensitization. Genomic instability can manifest several generations after exposure to many DNA-damaging agents, including ionizing radiation [71],[72],[73] and cause delayed non-apoptotic form of reproductive death in surviving cells.[74] Although the nature of the primary lesion(s) and the mechanism of this delayed chromosomal instability induced by radiation remain largely unknown, it has been speculated that some of the radiation-induced DNA lesions may trigger cellular processes, including gene deletions, eventually leading to delayed chromosomal instability.[71],[72],[73] A careful examination of the IC50 values and the MF values as well as delayed apoptosis (observed at 72 h after treatment) suggests that lesions induced by the ligand (mainly cleavable complexes) are not only capable of inducing genomic instability-linked non-apoptotic death, but are also capable of enhancing this effect, which may partly account for the synergistic cell kill observed in some (BMG-1 & U-87), but not in all cells. Hoechst-33342 has been shown to induce apoptosis by itself involving a number of metabolic perturbations, which appears to be mitochondria dependent, but p53 independent,[75] as observed in the present studies for the induction of apoptosis by hoechst as well as the combined treatment. Some of these perturbations include inhibition of TOP1 and interference in TATA binding protein leading to the stalling of RNA pol II involved RNA synthesis.[76] Interaction of lesions induced by hoechst-33342 and radiation resulting in the formation of complex irreparable damage may accentuate these processes resulting in enhanced apoptosis. Since the levels and activities of topoisomerases as well as repair capacities of cells differ considerably among different tumor cell lines, the induction of apoptosis may vary as observed in the present studies between the two glioma (BMG-1 & U-87) and squamous carcinoma (4451 & 4197) cell lines. Further, the induction of apoptosis has also been shown to be cyclin B1 mediated under certain conditions.[47] Therefore, differences in the endogenous as well as treatment-induced cyclin B1 levels may also be responsible for variations in the extent of apoptosis induction. The levels of bax and bcl-2 are important determinants of mitochondria dependent apoptosis, with the bax/bcl-2 ratio determining the release of Cytochrome c that activates caspases.[69] Preliminary observations show that the bax/bcl-2 ratios of 8-10 found in both the glioma cell lines (BMG-1 & U-87) were nearly two folds higher than the ratio (4-5) observed in the squamous carcinoma lines 4451 & 4197 (data not shown). Both ionizing radiation and hoechst-33342 induced cytotoxicity show cell cycle specificity. While G2 and M phase cells are more sensitive to ionizing radiation,[77],[78],[79] S-phase cells to a very great extent as well as G2 and M phase cells are sensitive to hoechst.[80] Therefore, heterogeneity in the responses among the cell lines could partly arise on account of differences in the cell cycle distribution at the time of treatment. However, in the present studies, the cell cycle distribution of the four cell lines at the time of treatment were essentially similar (data not shown) and may not be responsible for the differences in radiosensitization. Both hoechst-33258 and hoecsht-33342 are well known scavengers of free radicals[23],[81] and also suggested to quench long-lived DNA radicals,[82] thereby reducing the induction of DNA damage in solution[23],[83] and in cells under certain conditions.[29],[30] This suggests that hoehcst-33342 would protect cells against radiation-induced cytotoxicity, since DNA damage is primarily responsible for the cell death.[5] Indeed, reduced cytogenetic damage and enhanced animal survival have been reported in whole body irradiated mice when hoechst is administered before irradiation.[84] However, the ligand appears to be relatively inefficient in reducing the radiation-induced damage to the nucleotides under anoxic conditions,[82] suggesting that the ligand may not significantly reduce the damage induction due to variable oxygen tension among different cells and particularly the hypoxic cells in the tumor. On the other hand, inhibition of topoisomerases as well as chromatin structure dependent DNA repair would result in a higher level of residual DNA damage resulting in a higher level of cytogenetic damage, cell cycle perturbations, delayed apoptosis and genomic instability, thereby enhancing the cytotoxicity, as observed in the present studies [Figure - 1][Figure - 2][Figure - 3][Figure - 4][Figure - 5][Figure - 6]. Furthermore, interactions between radiation-induced DNA damage and lesions formed due to toposiomerase inhibition by the ligand would also enhance the cytotoxicity. Since tumor cells and tissues are often associated with reduced oxygen tension and elevated levels of topoisomerases, hoechst has a higher probability of inducing radiosensitization in tumor cells and tissues, although it has the potential to reduce the induction of DNA damage to a certain extent. Indeed, while a dose dependent radiosensitization has been observed in focally irradiated Ehrlich ascites tumors,[37] a significant protection in the form of enhanced survival and reduced bone marrow damage has been reported following whole body irradiation of the mice.[84] In this respect, therefore the ligand appears to be a promising radiosensitizer for application. However, issues related to the toxicity to the normal cells, particularly the proliferating bone marrow and other tissues in the form of mutagenic, teratogenic and clastogenic effects will have to be addressed before contemplating clinical studies. In this direction, we have recently shown that certain hoechst analogues like the dimethoxy analogue (DMA) and Trisbenzimidazole (TBZ) derivatives of hoechst-33258 are quite promising as radioprotectors and are not associated with the toxic effects seen with hoechst.[85],[86] However, the challenge will be to develop such ligands that exploit differences in the finer organization of the genome between tumor and normal cells, thereby affording differential radiomodification. ACKNOWLEDGEMENTS We thank Mr. Rohit Mathur and Dr Sudhir Chandna for useful discussions. These studies were supported DRDO, Ministry of Defence, Govt. of India, in the form of R & D projects (INM-280 & INM-301).
References
Copyright 2005 - Journal of Cancer Research and Therapeutics The following images related to this document are available:Photo images[cr05035f4.jpg] [cr05035t2.jpg] [cr05035f1.jpg] [cr05035f6.jpg] [cr05035f2.jpg] [cr05035f3.jpg] [cr05035t1.jpg] [cr05035_5.jpg] [cr05035f5.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}