 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
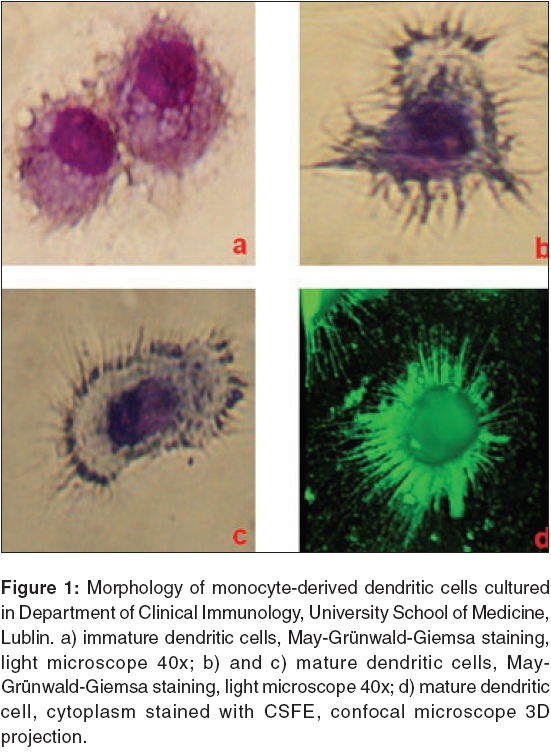
Journal of Cancer Research and Therapeutics, Vol. 2, No. 2, April-June, 2006, pp. 35-40 Review Article Dendritic cells heterogeneity and its role in cancer immunity Lin Kah-Wai, Tabarkiewicz Jacek*, Rolinski Jacek* Lviv National Medical University, 76 Pekarska Street, Lviv, Ukraine *Department of Clinical Immunology, University School of Medicine, Jaczewskiego 8, Lublin, Poland Code Number: cr06009 Abstract Dendritic cells are the most potent 'professional' antigen- presenting cells, with high ability of primary immune response initiation. Dendritic cells originate from bone marrow progenitors, which circulate in peripheral blood and subsequently give rise to immature dendritic cells, which reside in peripheral tissues. When dendritic cells encounter danger signals, they undergo differentiation and maturation; thereafter they migrate to lymphatic tissues, where they synapse with T-cells and initiate primary immune response. The immune response efficiency is determined by Th1/Th2 balance. Although dendritic cells represent a rare group of leukocytes, their functional and phenotypical heterogeneity confer a great challenge to immunologists. In this review, the myeloid and lymphoid development pathways of dendritic cells, are discussed. The heterogeneity of dendritic cells will be reviewed, based on their anatomical locations, phenotypes and functions. This section focuses on blood and lymphoid tissue dendritic cells. Subsequently, the roles of dendritic cells in the immunity of cancer and how cancer bypasses the dendritic cell-mediated immune responses, are discussed.Keywords: Dendritic cells, myeloid, lymphoid, cancer, immunity. Introduction Dendritic cells (DCs) are a rare population of leukocytes, with complex phenotypical and functional heterogeneity. Since DCs were first described by Steinman and Cohn in 1973, their role in immunity and potential in immunotherapy, have been intensely investigated. DCs are highly potent ′professional′antigen presenting cells (APCs), which are responsible for capturing, processing and presenting the antigen to T cells and thus priming the primary and secondary immune responses.[1],[2] DCs express high level of major histocompatibility complex (MHC) molecules, co-stimulatory molecules (CD40, CD80, CD86) and adhesive molecules (CD11a, CD15s, CD18, CD29, CD44, CD49d, CD50, CD54), which are a prerequisite for complete T cell activation.[3],[4] The ′professional′ APCs, such as DCs, macrophages and B cells, differ from ′non-professional′ ones, in their capacity of inducing the primary immune response. The ′non-professional′APCs, represented by most tissues, express low level of MHC molecules and do not express co-stimulatory and adhesive molecules. They induce tolerance, rather than effective immune activation.[5] DCs originate from CD34+ bone marrow progenitors. DC precursors circulate in the blood and subsequently give rise to immature DCs, which reside in peripheral tissues, such as Langerhans cells in epidermis and dermal DCs in dermis.[2],[6] Upon encountering antigens or other foreign stimuli, the subsequent production of local inflammatory chemokines will recruit more circulating DCs, via chemokine receptors.[6] DCs uptake and process the antigens in one of three pathways, namely MHC I, MHC II, or CD1 and then express them on their cell surfaces.[7] In response to Macrophage Inflammatory Protein 3 alpha (MIP-3α), DCs express chemokine receptors such as CCR6 (Chemokine Receptor 6), which may lead to a migratory process to the lymphoid organs.[6] Upon differentiation and maturation, DCs convert from an antigen-capturing mode, to a T cell priming mode.[8] The up-regulated MHC, co-stimulatory molecules and adhesive molecules, can effectively induce the formation of immunological synapses with antigen specific CD4+ T cells.[1],[2],[9] As a consequence, primary immune response is initiated and its efficiency is determined by Th1/Th2 (T helper 1/T helper 2) balance.[8] Despite intensive studies on DCs in the last decades, it is yet a myriad of unsolved questions, puzzling immunologists and biologists. These are important in the application of DCs in clinical practice, especially in the diagnosis and immunotherapy of malignancy, or autoimmune diseases. In this review, the myeloid and lymphoid development pathways of DCs, are discussed. The heterogeneity of DCs will be reviewed, based on their phenotype and function. Subsequently, the roles of DCs in immunity of cancer and how cancer bypasses the DC-mediated immune response, are discussed. Origin of Dendritic Cells DCs are rare and difficult to isolate, however, many studies are carried out on in vitro and in vivo systems, both in human and mice. Much knowledge on the DC′s developmental pathway has been obtained from the cell culture studies, but experimental results suggest that in vivo generation of some DCs, is unrelated to its counterparts, under different in vitro conditions.[10] The current model suggests, that DCs are divided into myeloid and lymphoid subpopulations, based on their developmental pathways. Functionally, myeloid DCs are immunogenic, while lymphoid DCs are rather tolerogenic. There is considerable evidence for the myeloid pathway of DC development. The peripheral CD14+ monocytes are common precursors of macrophages and DCs, in the in vitro condition.[11] Under the influences of GM-CSF (Granulocyte-Macrophage Colony Stimulating Factor) and IL-4 (Interleukin 4) in the 5 to 7 days culture, the CD14+ monocytes differentiate into immature DCs [Figure - 1]a. Further addition of pro-inflammatory mediators, such as TNF-α (Tumor Necrosis Factor-alpha), IL-1β , IL-6 and PGE 2 (Prostaglandin E2), for another 2 to 3 days, will induce phenotypical and functional mature DCs [Figure - 1]b-d. The CD14 expression is down-regulated and CD83, CD86 and MHC II, are up-regulated upon maturation.[12],[13] Other evidence of myeloid origin of DCs, may be derived from the studies of CD34+ precursors. The CD34+ precursors from bone marrow or peripheral blood can be differentiated, through at least two developmental pathways. These pathways exhibit common precursors, which have the potential capacity to generate either myeloid phagocytic cells (macrophages) or DCs (dermal DCs, Langerhans cells), implying the myeloid origin of DCs.[3],[10],[14],[15] Olweus et al have found a large subset of IL-3Rαhi DCs in the T cell-dependent areas of human lymphoid organs, derived from the precursors of myeloid origin, CD34+ IL-3Rαhi in blood and bone marrow.[16] The DCs derived from both CD14+ monocytes and CD34+ progenitors, expressed the myeloid-associated markers, CD33 and CD13.[3] The first evidence of lymphoid origin of DCs, was derived from the cell transfer study. When CD4low cells, the earliest precursors of mouse thymic T cells, are injected into a recipient, they not only give rise to the T cells, B cells and NK cells, but also DCs.[11],[17],[18] These CD4low derived DCs possess several lymphoid related markers, including CD8α, CD2, BP-1 and CD25.[3],[18] In in vitro studies, the mouse thymus CD4low cells differentiate into DCs and express mature DC markers, i.e. MHC II, CD11c, CD40, CD86, DEC-205 etc.[19],[20] The human bone marrow lin-CD34+CD10+ cells are able to generate T cells, B cells, NK cells and DCs. This indicates that the DCs developed in the lymphoid pathway.[21] The CD4+CD3-CD11c- plasmacytoid cells from human tonsil, blood, cord blood and thymus, differentiate into DCs phenotype. These cells express pre-T cell receptor a-chain and do not express myeloid markers and thus, are related to the lymphoid developmental pathway.[22] A recent study showed that bone marrow plasmacytoid DCs, upon viral infection, can differentiate into myeloid DCs. This result suggested the developmental flexibility and plasticity of DCs.[23] However, the developmental pathways of DCs are not fully understood and remain illusive and further investigations are needed to determine whether the DCs′myeloid and lymphoid routes, are mutually dependent or independent. Heterogeneity of Dendritic Cells DCs represent a very heterogeneous group of leukocytes. The heterogeneity of DCs, is reflected at four levels: the precursor populations, anatomical localization, function and final outcome of immune response.[6] Although many attempts to classify DCs subsets were carried out by phenotypical and functional analysis in the past few years, the complete model has not yet been established. DCs are widely distributed over the body, including Langerhans cells in epidermis, blood DCs, interstitial DCs in lymphoid and non-lymphoid tissues, interdigitating DCs, germinal center DCs and plasmacytoid DCs in lymphoid tissues etc.[1],[22],[24],[25],[26] Both immunogenic mature and tolerogenic immature phenotypes, are presented. However, the tolerogenic mature DCs, such as thymic DCs, induce tolerance in thymocytes by negative selection and pulmonary DCs induce response to antigen.[5],[27],[28] Lutz et al proposed the presence of an intermediate group, tolerogenic, semi-mature DCs, which express high level of MHC II and co-stimulatory molecules. In contrast to mature DCs, the semi-mature DCs express low level of cytokines, such as IL-1β , IL-6, TNF-α, and IL-12.[5] The analysis of human blood DCs are significantly hampered by its paucity (less than 1%) and the absence of specific markers. In addition, the inter-laboratory variation of purification protocols and monoclonal antibodies, result in different DC subset classifications.[26],[29] In most studies, immunophenotyping of blood DCs is often confined to a group of cells lacking lineage-specific markers (lin-) and possess high expression of HLA-DR (HLA-DRhi ).[26],[29] They do not express lineage markers, including CD8, TCR (T-cell Receptor) and CD90 for T-cell lineage; CD10 and CD20 for B-cell lineage; CD56 for NK-cell lineage; CD14 for monocyte lineage; CD15 and CD35 for granulocyte lineage; glycophorin A for erythrocyte lineage.[15] The circulating DCs are either precursor DCs, drained from bone marrow to peripheral tissues, or antigen-captured DCs, drained from peripheral to lymphoid tissues.[7] There are at least two subsets of DCs in peripheral blood: CD11c+CD123lo myeloid DCs and CD11c-CD123hi lymphoid DCs.[30] Ito et al defined three subsets of DCs: CD1a+CD11c+ monocyte-derived LC precursors, CD1a-CD11c+ monocyte-derived interstitial precursors and CD1a-CD11c- plasmacytoid DCs.[15] Later, Dzionek et al described novel markers for blood DCs: BDCA-1 (CD1c) and BDCA-3 (CD141), for two subpopulations of myeloid origin and BDCA-2(CD303) and BDCA-4(CD304), characteristic for lymphoid/plasmacytoid dendritic cells.[29] BDCA-1 (CD1c) is expressed on a subpopulation of human dendritic cells, which are of monocytoid morphology. These cells are CD4+, Lin-, CD11cbright , CD123dim , CD45RO+, CD2+ and express myeloid markers (CD13, CD33), as well as Fc receptors (CD32, CD64, FceRI). BDCA-2 is specifically expressed on human lymphoid (plasmacytoid) dendritic cells. Phenotyping of BDCA-2 positive cells, shows that these cells are CD4+, Lin-, CD11c-, CD123bright , CD45RA+, CD2- and expressing neither myeloid lineage markers like CD13 and CD33, nor Fc receptors. These cells are circulating in blood and are home to lymphoid and non-lymphoid tissues. BDCA-4 are expressed on CD11c-CD123hi lymphoid/plasmacytoid DCs and BDCA-3 is expressed on CD11c+CD123- myeloid DCs.[29] There are many studies on characterization of blood DCs with other markers, such as CD33, CD16, CD2, CD1, CD85, IL-3Rα, and so on.[16],[26] The study by MacDonald et al . on lin-HLA-DR+, where a wide range of monoclonal antibodies are used, defined five non-overlapping subsets of blood DCs: CD123, CD1b/c, CD16, BDCA-3 and CD34.[26] The CD16, CD1b/c and BDCA-3 DCs expressed CD11c, are suggested as myeloid origin, while the CD123+ and CD34+ DCs expressed CD11c, are suggested as lymphoid cells and hematopoietic progenitors, respectively. Although this study increased the specificity in categorizing blood DCs, the specific functions and stage of development were not defined.[26] There are many studies on subcategorizing DCs in mouse lymphoid tissues.[31],[32],[33] In contrast, there are few studies on human thymus DCs. In the study by Vansenabeele et al , two subsets of DCs are defined; more than 65% are CD11b-CD33lo CD45lo lymphoid-related plasmacytoid DCs and less than 35% are CD11b+CD33hi CD45hi myeloid-related DCs. The former groups are free of myeloid markers. The later group is represented by both mature and immature phenotypes.[22] The CD11b- DCs express TECT, which is indicated in early thymic development and migration of thymocyte subsets, by signaling through CCR9.[22] While the CD11b+ DCs express MIP-1a, an inflammatory chemokine signals through CCR1 and CCR5. The negative selection of self-reactive T lymphocytes, is dependent on IL-12 secretion by CD11b- cells. This may distinguish them from CD11b+ cells, which do not secrete IL-12, thus implying their functional difference.[22] The Vermare′s report showed similar results and three subsets are described, plasmacytoid DCs, immature myeloid DCs and mature interdigitating DCs.[34] In human tonsils, the interdigitating DCs, plasmacytoid DCs and germinal center DCs were defined in earlier studies.[15],[17],[24] Recently, five subsets of DCs are described, including three interdigitating DC subsets, plasmacytoid DCs and germinal center DCs.[24] As a result, the studies on lymphoid tissues indicated the heterogeneity of DC subsets with both myeloid and lymphoid origin, which are immunogenic and tolerogenic in function, respectively. Thymus is the central lymphoid organ and site of generation and selection of T-lymphocytes. Thymic DCs are heterogeneous and contain three subpopulations: a major subset derived from the precursors within thymus, a minor subset, presumably of extrathymic origin and plasmacytoid dendritic cells. Increasing evidence suggest that thymic cDC can cross-present self-antigens to developing thymocytes and play an important role in thymocyte negative selection and central tolerance induction. The role of thymic pDC is nowadays, still unclear.[35] The complexities of the heterogeneity of DCs, confer a great challenge to immunologists in characterization of DCs, because the different subsets of DC represent different developmental pathways, differentiation and activation stages and their correlation with their in vitro counterparts. The standard and practical model for clinical applications necessitate further investigation. How Dendritic Cells bypass immune responses in cancer The concept of immune surveillance, proposed that the clone of transformed cells are recognized and killed by the immune system, before they grow into tumor. However, experiments showed that the immune system, does not react against many tumor types.[36] Most tumor antigens are weakly immunogenic and the immune functions such as the antigen-specific T-cell response initiated by professional APC and immune regulation mediated by regulatory cells, often fail to function properly and thus cause the growth of tumors.[36],[37],[38] Many studies showed that the defect of dendritic cells, is one of the important factors, leading to the immune escape of tumor growth. The earlier studies of infiltration of dendritic cells into tumors, showed variable results in the context of prognosis.[39] The increased infiltration of DCs into esophageal carcinoma and hepatocellular carcinoma, is associated with good prognosis.[40],[41] In contrast, the studies on renal cell carcinoma showed no tumor regression by DCs.[42],[43] Mailliard et al suggested that infiltrating dendritic cells could promote T-cells survival or death, depending on their maturation stage and function.[44] They also described subpopulations of dendritic cells (NKDC, IKDC), which are able to destroy tumor cells, by direct interaction and secretion IFNs.[45],[46] However, the quantitative analyses of DCs, are not enough to explain the exact role of DCs in tumor regression. The more valuable and complete interpretations relied on the functional study on DCs, in which both systemic and local effect are important. The first event of the defect of DCs, may attribute to the production of tumor derived factors, that affect the DC′s differentiation and maturation processes. Many tumor derived cytokines that possess immunosuppressive activity, can impede the development of fully mature DCs, such as VEGF (Vascular Endothelium Growth Factor), M-CSF, IL-6, GM-CSF, IL-10, gangliosides and TGF-β (Transforming Growth Factor beta).[38],[47] The VEGF presents its role in tumor progression, by the formation of new blood vessels and inhibition of the maturation of DCs.[38],[48] Caux et al showed that IL-6 and M-CSF secreted by tumor cells, inhibit the differentiation of CD34+ progenitor cells into mature DCs and so, the antigen presenting function is disrupted.[49] Several studies showed that IL-10 blocks the up-regulation of co-stimulatory molecules and IL-12 production and thus, Th1 immunity is blocked and antigen-specific tolerance is induced.[50],[51],[52] The studies on gangliosides showed their inhibitory effect on development and function of CD34+ progenitors and monocytes derived from DCs.[52] Further characterizations of the immunosuppressive influence of cytokines on defect DCs maturation, are needed. The DC′s maturation is a control point in the initiation of immune responses. The immature DCs play a significant role in immune tolerance, by inducing Th2 response.[54] As a consequence of the maturation of defective dendritic cells, there is a decrease of mature and functionally competent DCs and an increase of immature DCs.[38] The decreased numbers of mature DCs are not able to initiate a tumor-specific immune response, while the increased numbers of immature DCs are functionally incompetent and induce the tolerance of T-cells. As a consequence, the tumor escapes from the surveillance of dendritic cells.[38] A recent study on lung cancer and breast cancer, showed significant decrease in DC populations.[55],[56] However, the defects of DCs are confined to myeloid DCs only and not to plasmacytoid, or lymphoid DCs.[57] The functional analysis on renal cell carcinoma and prostate cancer, showed that infiltrated DCs have lower allostimulatory activity, which is associated with the absence or low level of co-stimulatory molecules, CD80 and CD86.[58] The peripheral blood DCs of patients with breast cancer, expressed lower level of MHC II and co-stimulatory molecules.[59] The expression of impaired MHC II and co-stimulatory molecules, indicates disrupted T cell activation and subsequent immune activation. In the study by Almand, immature DCs in both lymph nodes and peripheral blood increased, but the ability to induce antigen-specific proliferation of autologous T-cells significantly decreased.[60] The knowledge of how defects of DCs cause progression of tumors, is clinically important. Although many studies have been carried out to define the mechanism of defects of DCs in cancer, many tumor types and the underlying molecular mechanisms are yet to be clearly defined. Conclusion The immunobiology of DCs developed rapidly in recent years, due to its great significance in clinical application. The specificity and plasticity of DCs, allowed DC-based immunotherapy to be effective against malignancy, autoimmunity and infectious diseases, in both the prophylactic and therapeutic approaches.[61],[62] However, many elementary problems in the experimental research have yet to be solved, especially relating to the heterogeneity of DCs and their role in various diseases. Addressing these problems would greatly stimulate the progression of clinical application. The knowledge of ontogeny of DCs and developmental pathways, would also have significant therapeutic value. The standardization of definitions of DC subsets, would be useful in clinical studies as well. Studies of the functional roles of DCs in various types of cancer and their mechanisms of escape from the DC-mediated immune response, can lead us to a better understanding of the immune system′s defensive response to differential tumor types, as well as the design of more effective therapeutic strategies.Acknowledgments The authors would like to thank Anna Michalak-Stoma, MD and Stan Moore, PhD for revising of article and Kamila Wojas, MSc for culture of photographed dendritic cells.References
Copyright 2006 - Journal of Cancer Research and Therapeutics The following images related to this document are available:Photo images[cr06009f1.jpg] |
| |||||||||

{kind=link}