 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
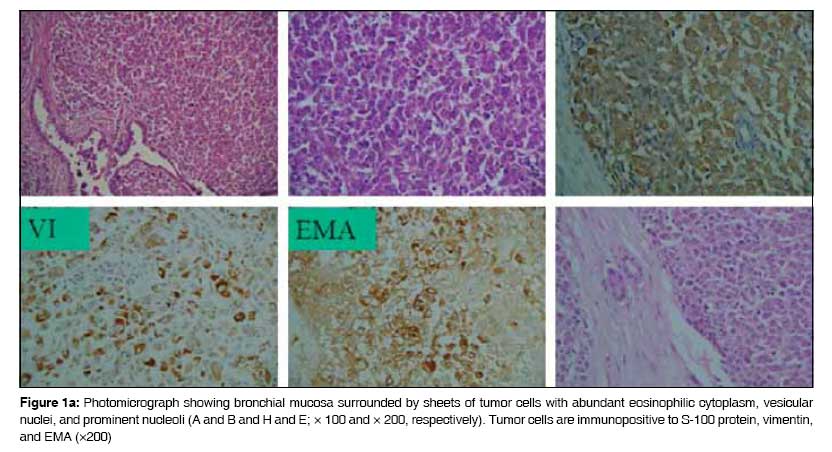
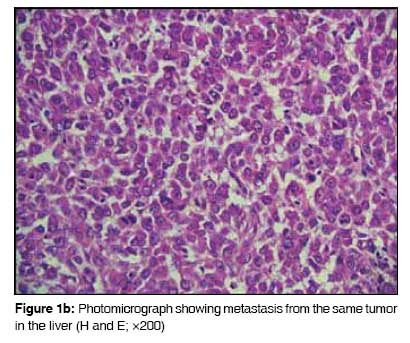
Journal of Cancer Research and Therapeutics, Vol. 5, No. 1, January-March, 2009, pp. 54-57 Case Report Rhabdoid variant of lung cancer: Clinicopathological details of a case and a review of literature Saini Gagan, Kumar Milind, Julka PramodK, Puri Tarun, Sharma Mehar, Rath GouraK Department of Radiotherapy and Oncology, All India Institute of Medical Sciences, New Delhi - 110 049 Code Number: cr09014 Abstract Primary rhabdoid tumor of lung is a rare histological and clinical entity. Lung tumors with rhabdoid features have been included as variants of large-cell carcinoma in the 1999 World Health Organization (WHO) classification of lung tumors. A large-cell carcinoma with a rhabdoid phenotype (LCCRP) is unusual, with only 38 cases reported till date. We report the clinical details of one such case that was treated with pneumonectomy and adjuvant chemotherapy. We also present a review of the literature. To identify relevant articles, we searched PubMed, Ovid, and IngentaConnect databases using the key words 'rhabdoid,' 'lung cancer,' and 'primary rhabdoid tumor of lung.'Keywords: Carcinoma lung, docetaxel, rhabdoid Introduction Primary rhabdoid tumor of lung is a rare histological and clinical entity. Its histological and biological characteristics have not been fully documented. Lung tumors with rhabdoid features were included as variants of large-cell carcinoma in the 1999 WHO classification of lung tumors. [1] We located 38 case reports of primary rhabdoid tumor of the lung in the English literature [Table - 1]. These tumors are characterized histologically by the presence of typical rhabdoid cells, which are large cells with abundant cytoplasm and eccentric nuclei with central macronucleoli; in addition, these cells show a rounded eosinophilic cytoplasmic inclusion. These tumors behave aggressively and, in most of the reported cases, the clinical course has shown rapid progression and early death. Despite a thorough search of the literature we could not find another case report from India. We present our experience of treating a case of primary rhabdoid tumor of the lung.Case Report A 36-year-old nonsmoker lady presented to our institute in March 2003 with three to four episodes of hemoptysis. She did not have productive cough, fever, chest pain, or dyspnea. She was pregnant and in the third trimester at the time. Computed tomography (CT) scan of the chest showed a mass of size 5 x 5cm in the upper lobe of the left lung, which was central and abutting the mediastinal vascular structures. No mediastinal or hilar lymphadenopathy was evident radiologically. Bronchial biopsy was strongly suspicious of malignancy. The complete metastatic workup was normal. Medical termination of pregnancy was done in April 2003. She underwent left pneumonectomy in May 2003. At surgery, a left lung mass was seen that was adherent to the main pulmonary artery; no pleural effusion or deposits were observed. Postoperatively, the pathology report showed a 6 x 5cm tumor that under microscopic examination [Figure - 1a & b] was markedly cellular, with the cells arranged in lobules separated by fibrocollagenous septae. The tumor cells were oval to polygonal in shape, with abundant cytoplasm and a vesicular nucleus; some of the nuclei had a prominent nucleolus with typical globules. Immunohistochemical staining [Figure 1a] showed positivity to S-100 (focal), vimentin, and EMA. It was negative for LCA, HMB-45, and CEA. A diagnosis of large-cell carcinoma with rhabdoid phenotype (LCCRP) was made. The resection margins of the bronchus and lymph nodes were free of tumor. The patient received adjuvant chemotherapy with intravenous (IV) paclitaxel (175 mg/m 2 ) on Day 1 and IV carboplatin AUC5 on Day 2, 4-weekly for 6 cycles from August 2003 to January 2004. She remained free of disease clinically, as well as on imaging, till November 2005. Follow-up CT scans of the chest and abdomen done in November 2005 revealed solitary liver metastases in segments 2 and 3, with no evidence of disease in the chest. Fine needle aspiration cytology (FNAC) from the liver lesion revealed metastatic, undifferentiated carcinoma. She underwent left lateral hepatectomy in January 2006. Postoperative histopathology confirmed metastatic rhabdoid tumor with negative margins of resection [Figure 1b]. Postsurgery positron emission tomography (PET) scan done in February 2006 showed mild increase in fluorodeoxyglucose (FDG) uptake along the left lateral margin of the liver, which was reported as most probably being a postoperative change. She received 3-weekly adjuvant chemotherapy with six cycles of IV docetaxel (75mg/m 2 ) on Day 1 from February 2006 to June 2006. A postchemotherapy PET scan was done in July 2006 and showed no evidence of active disease. A repeat PET scan in February 2007 showed a sustained remission [Figure - 2]. She developed recurrence at the mediastinal lymph nodes in December 2007 and is now being treated with taxane-based chemotherapy. The latest PET scan, which was done in April 2008, showed significantly decreased avidity of FDG uptake. Discussion Malignant rhabdoid tumors were described first in the kidney by Beckwith et al . [2] in 1978 and were thought to be associated with aggressive characteristics. The term 'rhabdoid' was chosen because the tumor cells were similar in appearance to the cells of rhabdomyosarcoma. These tumor cells were large cells, with abundant cytoplasm, a large eccentric nucleus, and a central macronucleus; additionally, these cells were characterized by the presence of a rounded eosinophilic cytoplasmic inclusion within the cytoplasm. Tumors with such rhabdoid appearance have been described in many extrarenal sites. There is, however, a lack of pathological uniformity in these cases and therefore the concept of extrarenal rhabdoid tumors has not been accepted as a pathological diagnostic entity by many. [3] Metastases to the lung from rhabdoid tumors of the kidney and extrarenal rhabdoid tumors is a relatively common finding, and therefore before making a diagnosis of a primary rhabdoid tumor of the lung, clinical, pathological, and radiological investigations must be done to exclude a primary tumor located elsewhere. [4] Primary rhabdoid tumor of the lung has been infrequently reported. The first report of a rhabdoid tumor in the lung was by Colby et al . in 1995 [5] who described a neuroendocrine carcinoma with rhabdoid phenotype. This patient was also included in the series of six cases that was subsequently reported by Cavazza et al . in 1996. [6] Cavazza et al ., in their case series, proposed a diagnostic criterion for lung tumor with a rhabdoid phenotype: at least 10% of the tumor cell population must consist of rhabdoid cells. However, they did not give a detailed explanation or describe the methodology. To the best of our knowledge, only 38 cases have been reported in the English literature to date [Table - 1]. The largest series to date has been of 11 patients, which was by Tamboli et al . in 2003. [4] Microscopically the tumor cells in primary rhabdoid tumor of lung are usually high grade or poorly differentiated. Most of these tumors show a combined mixed morphology, with a rhabdoid component and an epithelial or mesenchymal component. They usually express epithelial markers like cytokeratin and epithelial membrane antigen, nonmuscle mesenchymal cell markers like vimentin, as well as various neuroendocrine markers [Table - 2]. These tumors have been analyzed for a variety of markers and show heterogeneity in expression of various markers. Ultrastructurally, the hallmark of the rhabdoid cell is a large paranuclear, hyaline, often globular, eosinophilic cytoplasmic inclusion that sometimes causes nuclear indentation. This inclusion ultrastructurally is composed of cytoplasmic intermediate filaments that are arranged in a concentric whorl-like pattern. [4] The globular eosinophilic inclusions are immunoreactive for vimentin and that, in some cases, is the only immunopositive reaction seen in a rhabdoid tumor. This presence of this feature can help the pathologist reach this diagnosis. Cavazza et al . [6] have commented that the histological differential diagnoses of tumors of the lung with a rhabdoid phenotype includes a variety of malignant primary and metastatic neoplasms, e.g., mucinous adenocarcinoma with signet-ring cells, malignant melanoma of lung, rhabdomyosarcoma, epitheloid angiosarcoma, and plasmacytoma. In an important review article by Wick et al . [7] in 1995, the term 'composite extrarenal rhabdoid tumor' was put forward. They suggested that 'rhabdoid tumors' are a heterogeneous group of lesions with dissimilar lineages of differentiation with dissimilar histogenesis. They reported the existence of totipotent stem cells in many tissues, which were capable of epithelial, mesenchymal, or mixed differentiation, and commented that the rhabdoid phenotype may represent 'a common endpoint of clonal evolution in tumors of clearly differentiated origins.' In an analysis by Miyagi et al ., [8] proliferative cell nuclear antigen (PCNA) was strongly demonstrated in cases of rhabdoid lung tumors. In two of three cases, 70-90% of cells showed PCNA, which probably indicates the highly proliferative Nature of the tumor. The rhabdoid variant of large-cell carcinoma of lung was included in the WHO classification of lung cancer [1] in 1999 and hence can be considered an established pathological entity. Clinically, rhabdoid tumors are aggressive tumors with most cases presenting at advanced stages. [4] The tumor carries a bad prognosis due to its rapid and fatal progression. In our case, the tumor was abutting the pulmonary artery and hence the patient had hemoptysis, which probably led to early diagnosis while the tumor was still at an operable stage. She has now survived for 5 years after diagnosis. The first relapse occurred 1 year after diagnosis and, post liver resection, she was disease free for 24 months. After this there was a second relapse, which has shown response to retreatment with taxanes. In short, our case has shown satisfactory results with timely surgery and adjuvant chemotherapy with rigorous follow-up. Rhabdoid tumor of lung, being a rare entity, is usually treated similarly to non-small-cell carcinoma of the lung. We should recognize the poor prognosis associated with this histology; a timely and aggressive multimodality approach is needed to achieve long-term disease control.[17] References
Copyright 2009 - Journal of Cancer Research and Therapeutics The following images related to this document are available:Photo images[cr09014f2.jpg] [cr09014f1b.jpg] [cr09014f1a.jpg] [cr09014t2.jpg] [cr09014t1.jpg] |
| |||||||||

![[Table - 1]](/showimage?cr/photo/cr09014t1.jpg){kind=link}
{kind=link}
{kind=link}
![[Figure - 2]](/showimage?cr/photo/cr09014f2.jpg){kind=link}
![[Table - 2]](/showimage?cr/photo/cr09014t2.jpg){kind=link}