 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
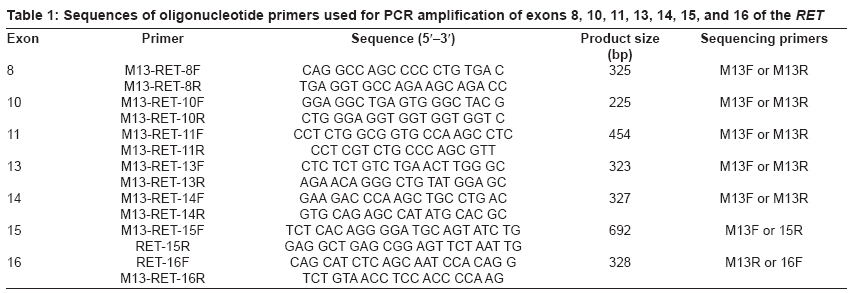
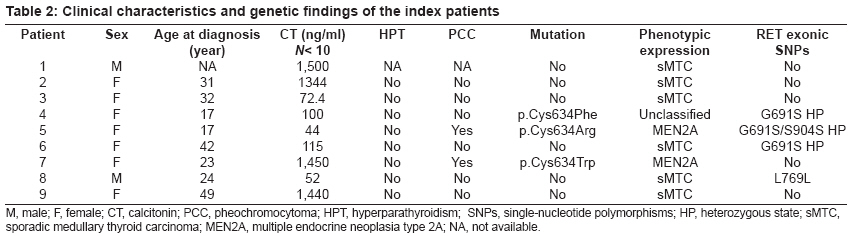
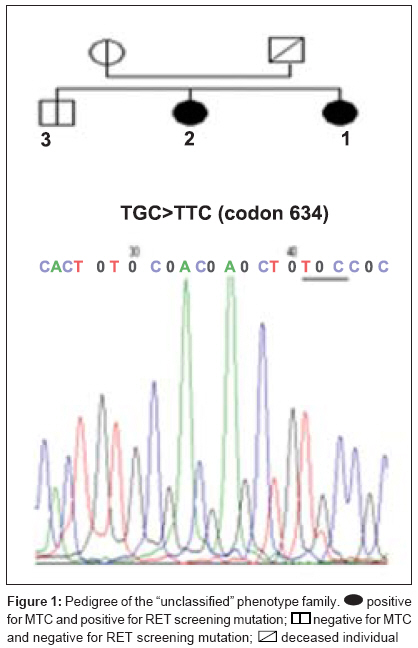
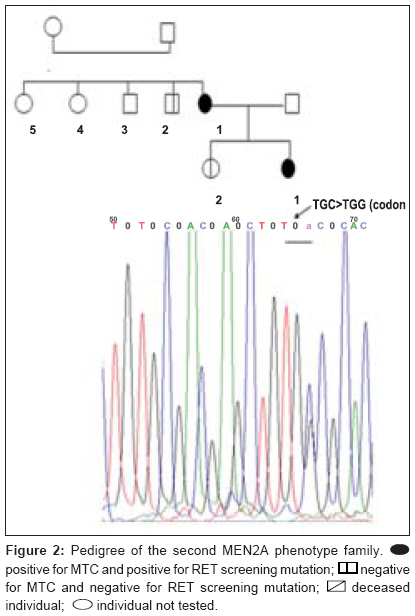
Journal of Cancer Research and Therapeutics, Vol. 5, No. 3, July-September, 2009, pp. 198-202 Original Article RET genetic screening in patients with medullary thyroid cancer: The Moroccan experience Abdelhakim Ainahi, Anne Barlier, Mohamed Kebbou, Nadia Benabdeljalil, Mohammed Timinouni, Taoufiq Fechtali, Catherine Roche, Said ElAntri Laboratoire d'Hormonologie et Marqueurs tumoraux Code Number: cr09048 PMID: 19841562 DOI: 10.4103/0973-1482.57126 Abstract Background : Germline RET gene mutations are well known to be the genetic causes of multiple endocrine neoplasia type 2 (MEN2) and may be identified by genetic screening.Aim : The purpose of the present study was to screen nine MTC patients for RET sequence changes. Materials and Methods : In this study, our sample was composed of 30 individuals: 9 index patients with medullary thyroid carcinoma (MTC) corresponding either to 3 subjects with clinical evidence of MEN2, 6 with apparently sporadic MTC (sMTC), and 21 relatives have been investigated for RET mutations. After DNA extraction from peripheral blood leukocytes, RET exons 8, 10, 11, 13-16 and exon/intron boundaries were analyzed by direct PCR sequencing. Results : Three different known RET germline mutations in exon 11 (codon 634), p.Cys634Arg (c1900 T→C) (de novo case), p.Cys634Phe (c1901 G→T), p.Cys634Trp (c1902 C→G), were detected in three individuals with MEN2 phenotype. Of the 21 relatives, 2 cases presented mutation. Among the six probands with sMTC, none was found to carry mutation. There was no difference between RET polymorphisms detected among both MEN2 and sMTC patients. Conclusions : These preliminary data suggest that the RET mutation spectra observed in Moroccan patients with MEN2 are similar to those previously reported in other countries. Keywords: Calcitonin, MTC, RET, MEN2A, polymorphism Introduction Medullary thyroid carcinoma (MTC) originates from para-follicular C cells and can be sporadic (75%) or hereditary (25%). [1] The familial form is an autosomal dominant inherited disease characterized by the presence of MTC and other endocrine tumors such as pheochromocytoma (PCC) and/or parathyroid adenoma (HPT) in multiple endocrine neoplasia type 2A (MEN2A), and PCC, ganglioneuromatosis, mucosal neuromas and/or skeletal abnormalities in MEN2B. Familial MTC (FMTC) is characterized by the familial occurrence of MTC without other lesions. [1],[2] Moreover, a MEN2 family with less than four members with only MTC and screened negative for PCC and HPT is referred to as "unclassified" or "other" according to the International RET Mutation Consortium criteria. [3],[4] Germline RET hotspot activating mutations affecting exons 8, 10, 11, 13, 14, 15, and 16 have been found to be associated with MEN2 and FMTC. [2],[3],[4] On the other hand, several RET single-nucleotide polymorphisms (SNPs) have been described in MTC patients as well as in the general population. [5],[6] It still is a matter of debate to which extent neutral sequence variants (polymorphisms) could have interacting, predisposing or modifying roles in the pathogenesis of MEN2 or sporadic MTC . [5],[6] Biochemical screening for early MEN2 in at-risk individuals is performed by testing basal and stimulated serum calcitonin (CT) concentration. [7],[8] Recently, presymptomatic identification of RET mutation carriers has had a great impact on the diagnosis and the management of MEN2 disease at an early stage. [8],[9] The aim of this study was to identify RET hotspot germline mutations and polymorphisms in MTC patients and their relatives. Materials and Methods Patients and their family members The classification of patients was done based on clinical results according to the International RET Mutation Consortium definitions. [3],[4] The population study was composed of 30 subjects, 9 index patients (2 MEN2A, 1 "unclassified", and 6 suspected sMTC), and 21 relatives. Median age at diagnosis was 20 years (MEN2) and 38 years (sMTC). All nine probands were clinically affected by MTC and were subjected to thyroidectomy. The anatomopathological analysis confirmed the MTC diagnosis. One patient died of the disease. Endocrinological testing Basal plasma CT concentrations were measured with a commercially available two-site immunometric assay (Elsa-hCT, Cis-BioInternational, Gif sur Yvette, France). A basal CT value up to 10 pg/ml was considered normal according to the data of the French Calcitonin Tumors Study Group. All probands were screened for the presence of PCC by HPLC catecholamine urinary level measurement methods (Chromosystems, Mόnchen, Germany). HPT screening was performed by measuring serum calcium (Vetros 250, Johnson and Johnson, USA) and parathyroid hormone levels (Elsa-PTH, Cis-BioInternational, Gif sur Yvette, France). RET mutation analysis After signed informed consent, genomic DNA was isolated from whole-blood samples and was frozen until testing in the Laboratory of Biochemstry and Molecular Biology at the Hospital Conception (Marseille, France). RET hotspots (exons 8, 10, 11, 13-16) were amplified using flanking regions′ primer setting [Table - 1], universal primer M13F (5´-TGT AAA ACG ACG GCC AGT-3´), and M13R (5´-CAG GAA ACA GCT ATG ACC-3´) according to the manufacturer′s protocol. [10] The genomic DNA was amplified using the Touchdown (TD) PCR assay [11] and HotStarTaq PCR kit (Qiagen, Courtaboeuf, France) in a final volume of 50 μl containing PCR buffer 1 x contains 1.5 Mm MgCl 2 , Q-Solution 1 x, 200 μM dNTPs, 2.5 U of HotStarTaq DNA polymerase and 0.5 μM of each primer. In a thermal cycler (The Applied Biosystems′ 2720 Thermal Cycler), TD amplification was performed with an initial step of 95 °C for 15 min , followed by 3 cycles of 95°C for 30 s, annealing temperatures starting at 66 °C for 45 s (decreasing by 2 °C/cycle), and 72 °C for 1 min for extension. This step was followed by 26 cycles of 95 °C for 30 s, 56 °C for 45 s, 72 °C for 1 min, and a final extension at 72 °C for 10 min. The amplified DNA was analyzed on 2% agarose Tris-borate-EDTA gel electrophoresis and purified using minispin columns (Millipore, Moslheim, France). Direct sequencing of the seven exons was carried out using Dye Terminator Cycle Sequencing Quick Start Kit (Beckman Coulter, CA, USA) on Ceq 8000 sequencer (Beckman Coulter) automated sequencer according to the manufacturer′s instructions. Results We examined 9 index cases suffering from MTC (mean age, 37.8 ± 15.8 years) and 21 relatives, totalizing 30 individuals. As shown in [Table - 2], MEN2 phenotype was restricted to mutations in exon 11 at codon 634 but there were different amino acid substitutions (replacements of cysteine with arginine, tyrosine, or tryptophan). However, RET germline mutation screening was negative in all suspected sMTC patients. Furthermore, RET hotspot sequencing revealed polymorphic variants at codon 691 (exon 11), 769 (exon 13), and 904 (exon 15) both in sMTC and MEN2 groups. The first MEN2A phenotype family was already described in a previous report. [12] The index patient (Patient 5 [Table - 2]) was a 18-year-old female. The relatively young age of PCC and MTC presentation suggested a potential syndrome and the following evaluation showed the presence of the germline mutation p.Cys634Arg (c1900 T;C), while the genetic screening of the remaining family members (the parents, three sisters, and one brother) was found negative suggesting a de novo index case. In the "unclassified" phenotype family, the index patient (Patient 4 [Table - 2]), a 24-year-old girl, harbored the germline mutation p.Cys634Phe (c1901 G;T) [II-1, [Figure - 1]]. During the genetic screening, the proband′s sister was identified as a gene carrier at an age of 21 years. The former was clinically affected [II-2, [Figure - 1]] and showed increased basal CT (2025 pg/ml). She underwent total thyroidectomy. Pathology analysis was consistent with MTC. The remaining family members (the mother, one sister, and one brother) were negative for the mutation. However, the family history revealed that the proband′s father died due to metastatic disease many years prior to genetic testing. Until now, none of the two gene carriers from this family have had biochemical evidence of PCC or HPT. In the second MEN2A phenotype family, the index patient (Patient 7 [Table - 2]) was a 30-year-old woman. She was asymptomatic until the age of 25 years when a nodule in the right lobe of the thyroid gland was discovered. Total thyroidectomy was performed and the histological report showed the presence of MTC. At the time of diagnosis, PCC was also detected but no evidence of HPT was found. By DNA sequencing, the germline mutation p.Cys634Trp (c1902 C;G) [II-1, [Figure - 2]] was detected in this index case. Genetic screening of the family identified one positive carrier (the proband′s daughter). The clinically asymptomatic daughter, 11-year-old [III-1, [Figure - 2]], was a gene carrier with a moderately high basal CT level (13.88 pg/ml) and no biochemical evidence of PCC or HPT at this time. Based on those results, she was submitted to prophylactic thyroidectomy. Discussion Over the last 10 years, different RET mutations have been well characterized, especially after the introduction of RET genetic screening in the workup of all patients with MTC, of both hereditary and apparently sporadic type. [3],[4] Moreover, there are numerous new indices indicating a link between the known RET mutations from those classically associated with MTC to the ones associated with MEN2A or the opposite, showing a great variability of the mutational spectrum and the possible modulatory effect of the SNPs on the clinical expression. [13] Recently, genetic diagnosis for MEN2 disease has been available in Morocco. [12],[14] However, this is the first comprehensive report of RET mutation and polymorphism screening in a Moroccan population. In this research, we detected RET germline mutations with a high sensitivity analyzing exons 8, 10, 11, and 13-16. Mutations were identified in three (33%) cases; these results are in line with the data stating that up to 25% of MTC cases reported in several MTC families worldwide. This result confirms that RET mutations are highly conserved despite ethnical variations and environmental factors. [15],[16],[17] Our results correlate with the findings of the International RET Mutation Consortium showing that the most frequent mutations were found in exon 11 at codon 634. [16],[17] According to different series, the presence of a specific mutation at codon 634 has been associated with PCC and/or HPT; we observed this association in only two MEN2A cases. [16],[17] However, the small number of our samples does not allow us to draw conclusions on phenotype/genotype correlations. The p.Cys634Phe mutation, which is usually described in the literature as the cause of MEN2A, [3],[4] was found in the "unclassified" case. This fact could be explained either by the misclassification of MEN2A with low penetrance of PCC or by the possible influence of RET polymorphisms or by other modifier genes that protect this case from the development of PCC. [18],[19],[20] In this report, the early detection of mutation carrier was successful in two cases subjected to prophylactic thyroidectomy. Thus, genetic screening provided an important decision-making tool to guide clinical treatment in those cases. The decision to perform thyroidectomy on these young, at-risk carriers was due to the presence of RET 634 germline mutation and family history as well. Our results can be considered a good example of the clinical impact of early RET screening mutations on asymptomatic MTC carriers. [21] On the other hand, several authors suggest that a routine application of RET testing should be included in all cases of apparent sMTC because individuals could have germline mutation in the peripheral blood DNA, commonly associated with hereditary MTC. [21],[22],[23] In our samples, none of the six cases with sMTC revealed mutation, thus discarding inheritable disease. Mutations in other domains of RET or molecular alterations in other genes might be involved in the genesis of sMTC. [18],[19] It is also useful to mention that there was a difference in the mean age at diagnosis of the MTC between the sporadic and inherited groups of the family: 38 years (clinically sporadic cases) versus 20 years (clinically MEN2 cases). The inherited MTC form was associated with a better outcome due to the young age at the time of diagnosis. A similar association between poor prognosis and sporadic MTC has been found by other authors. [22],[23],[24] The identification of RET mutations, responsible for MEN2 syndrome, gives us the opportunity to find mutation carriers among at-risk family members and simplifies the management of kindred having this disease. Taking into considering the very small number of our observations, we have showed that the genetic characteristics of MTC among Moroccan patients with inherited or sporadic MTC are similar to those already described in several MTC families worldwide. Acknowledgments We thank Ms Germanetti Anne Laure and Dr. Sylvie Monique for their valuable technical assistance. This work was supported in part by the Oncogenetic Network from French Ministry, the assistance Publique des Hτpitaux de Marseille, and the International Atomic Energy Agency (IAEA). References
Copyright 2009 - Journal of Cancer Research and Therapeutics The following images related to this document are available:Photo images[cr09048t1.jpg] [cr09048f1.jpg] [cr09048t2.jpg] [cr09048f2.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}