 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
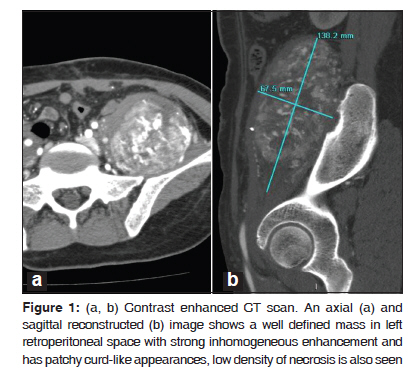
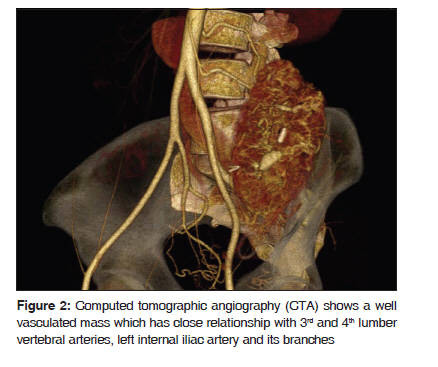
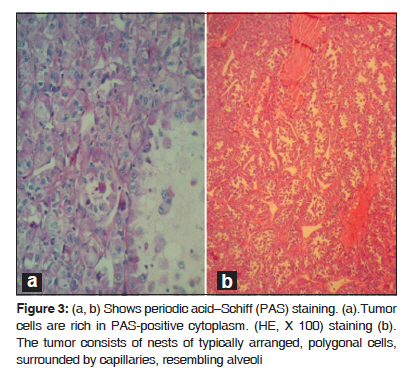
Journal of Cancer Research and Therapeutics, Vol. 6, No. 1, January-March, 2010, pp. 117-119 Case Report Alveolar soft part sarcoma of the retro peritoneum Fan Yu Xin, Netra Rana, Zhang Ming, Yu Bo Lang Department of Radiology, First Affiliated Hospital of Xi’an Jiaotong University (School of Medicine), Xi’an 710061, P.R.China Code Number: cr10029 DOI: 10.4103/0973-1482.63544 Abstract Alveolar Soft Part Sarcoma (ASPS), also called Alveolar Soft-Tissue Sarcoma, is a rare type of soft-tissue neoplasm with a poor long term prognosis. Such tumors originating in the retro peritoneal space are extremely rare. In this article we discuss a 34-year-old woman who was referred to our hospital with an increasing mass in her left lower abdomen. Ultrasonography and conventional Computed Tomography revealed a large hard mass occupying the left retroperitoneal space with a clear border. The pathological diagnosis was ASPS.Keywords: Alveolar soft-part sarcoma, immunohistochemical, myogenic, pseudo alveolar, retro peritoneum Introduction Alveolar Soft Part Sarcoma (ASPS) was first described by Christopherson and colleagues in 1952. [1] The tumor cells are rich in cytoplasm, containing periodic acid-Schiff- and diastase-positive granules and typical crystals. Mitoses are infrequent; the origin of ASPS is considered to be myogenic with a pseudo alveolar pattern formed by aggregates of large granular cells surrounded by vascular channels that mimic the alveolar pattern of respiratory alveoli. [2] Case Report A 34-year-old woman was referred to our hospital with an increasing mass in her left lower abdomen. Six month ago, the mass was walnut-sized with no significant pain and was not responsive to an anti-inflammatory therapy. Preoperative (USG) and Computed Tomography (CT) revealed a large hard mass occupying the left retroperitoneal space. The contrast enhanced CT showed a well defined mass (138.2 mm × 77.0 mm ×67.5 mm) in the left retroperitoneal space [Figure - 1] with inhomogeneously enhanced, patchy, curd-like appearance. A low density of necrosis was also observed. Computed Tomographic Angiography (CTA) indicated a well vasculated mass that had close relationships with 3 rd and 4 th lumber vertebral arteries, left internal iliac artery and its branches [Figure - 2]. Intra operative findings revealed a large, encapsulated, well vascularized tumor located in the left great psoas muscle. The mass was well dissected, no adhesions were found between the neoplasm and peritoneum. While omentum majus was adherent to the parietal peritoneum, no ascites were present. In gross pathological examination, the mass was delicate and jelly-like; the contents of mass were dark-yellow to dark-red in color with little hemorrhage. The tumor cells were rich in cytoplasm, containing Periodic Acid-Schiff (PAS), diastase-positive granules, and typical crystals [Figure - 3]a and b. The immunohistochemical (IHC) markers; Vimentin (VIM), Desmin (DES), NSE, S-100, were positive, while Myoglobin (MYO) and CD68 were non reactive. Histopathologic examination of the specimen revealed ASPS of the retro peritoneum. Discussion ASPS is a rare type of soft-tissue neoplasm with poor long term prognosis. The tumor presents itself with a pseudo alveolar pattern formed by aggregates of large granular cells surrounded by vascular channels that mimic the alveolar pattern of respiratory alveoli. [1] It is also known as malignant nonchromaffin paraganglioma and malignant organoid granular cell myoblastoma. The tumor accounts for approximately 0.5-1% of soft-tissue sarcomas and 0.1% of sarcomas of the head and neck. The tumor affects primarily younger patients with a female preponderance; the peak age of incidence is between 15 and 35 years. ASPS occurs most often in the soft tissues of the pelvis and the lower limbs and is very rare in the retroperitoneal space. [2] The histogenesis of the tumor remains uncertain; they are negative for neuroendocrin and epithelial markers. Myogenic or neural crest origin has been reported on the basis of electron microscopic findings. The tumor cells are rich in cytoplasm, containing periodic acid-Schiff and diastase-positive granules and typical crystals. These granules contain monocarboxylate transporter 1 (MCT1)-CD147 complexes. The nuclei are round or oval, have an irregular chromatin pattern, and their nucleoli are clear, without frequent mitoses. [3],[4] Differential diagnosis of ASPS revolves around neoplasms that may show nested or organoid patterns of growth, and cells with abundant eosinophilic cytoplasm. Renal cell carcinomas, adrenal cortical carcinomas and hepatocellular carcinomas may mimic ASPS because they share some morphological features, and common genetic substratum by virtue of their abundant eosinophilic to clear cytoplasm. Pang et al. [5] have reported the expression of ASPL-TFE3 fusion transcripts in paraffin-embedded tumor tissues to serve as a useful molecular marker in the diagnosis of ASPS, and also helpful in elucidating the underlying pathogenesis. Saito et al. [6] and Toguchida et al. [7] have reported the possibility of p53 gene mutations associated between tumor progression and genetic alteration in ASPS and other types of sarcoma. Local recurrence rate is not high; however, distant metastasis is common where lungs are the most frequently affected organs. However, Kayton et al. [8] have reported residual and local recurrence. Complete surgical resection is the mainstay of therapy. The utility of adjuvant treatment with chemotherapy or radiotherapy remains uncertain. Postoperative adjuvant radiotherapy and chemotherapy has been thought to be effective on reducing the risk of local recurrences and distant metastases. Regarding the imaging features, ASPS tends to be on par or at slightly higher signal intensity than the skeletal muscles on a T1-weighted MRI, and has high and heterogeneous signal intensity on a T2-weighted MRI. On unenhanced CT scans, ASPS generally displays an attenuation that is less than or equal to that of the surrounding muscle. Tumor margins on CT images can vary from well-defined to infiltrating. Central necrosis can be seen in up to 75% of the cases. ASPS shows strong enhancement on contrast enhanced CT and MR images. Small areas that fail to have enhancement may represent tumor necrosis. The presence of a large soft-tissue mass associated with large peritumoral vessels is strongly suggestive of ASPS. [4],[9] Ultrasound reveals a variable echo pattern, while Color Doppler Ultrasonography (CDUS) shows prominent vascularity within the tumor. The USG pattern is heterogeneous hypoechogenicity, infiltrated margins, ovoid contour, solid content, moderate size, marked hypervascularity on CDUS, and low resistive index (RI). [10] Conclusion ASPS is a rare type of soft-tissue neoplasm with a poor prognosis in the long term, and occurs most often in young adults with a female preponderance and less frequently in children. It commonly involves the muscles and soft tissues, particularly those of the lower extremities (buttocks, thighs and legs). It may arise in the upper extremities, head and neck regions, especially in a child; but could also have extra muscular localizations, in the female genital tract, the trunk, the mediastinum, or the retro peritoneum. Metastases are frequent, mainly in the lungs, bones, and the brain. Complete surgical resection is the mainstay of therapy; depending upon the number of metastases, chemotherapy with or without radiation, or surgery may be chosen. References
Copyright 2010 - Journal of Cancer Research and Therapeutics The following images related to this document are available:Photo images[cr10029f1.jpg] [cr10029f2.jpg] [cr10029f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}