 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
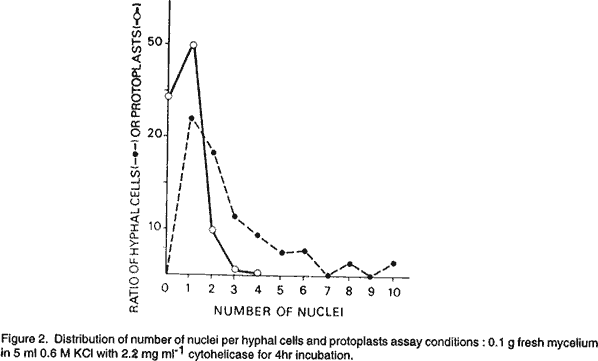
African Crop Science Journal, Vol. 9. No. 2, pp. 351-358 Isolation and regeneration of protoplasts from mycelium of Fusarium solani F. ELBOUAMI (Received 10 October, 1999; accepted 17 December, 2000) Code Number: CS01016 INTRODUCTION Genetic studies of the genus Fusarium are lacking probably because many species are entirely asexual. The only tool available for genetical analysis in these fungi is the parasexual cycle, but a clear demonstration of this cycle is lacking in many of these species. Nutritionally-forced heterokaryosis between complementary auxo-trophes has been reported (Burnett, 1984). Haploid recombinants were occasionally recovered from heterokaryons suggesting a parasexual mechanism in Fusarium species, but data were very incomplete. Protoplast fusion in filamentous fungi has proved to be a highly efficient procedure for obtaining heterokaryons and making the parasexual cycle readily available in many species (Anne, 1983; Daboussi and Gerllinger, 1992). We used this technique to investigate the occurrence of parasexuality in Fusarium solani. However, the development of a suitable method for generating sufficient quantities of protoplasts appeared to be pre-requisite in order to achieve the fusion (Peberdy, 1979). Thus, experiments were carried out to examine the influence of several parameters on protoplast release in an attempt to maximise the protoplast yield. In this paper, we report on an efficient method for isolating protoplasts and their regeneration to mycelial form. MATERIALS AND METHODS Organisms and culture conditions. Fusarium solani, a wild-type strain, was used to obtain protoplasts. Cultures were maintained on potato dextrose agar (PDA). Mycelium for the production of protoplasts was grown on PDA supplemented with 0.1% (w/v) yeast extract (PDAY). Trichoderma harzianum and Streptomyces venezuela, RA were kindly provided by Professor J.F. Peberdt (University of Nottingham, UK). The former was maintained on PDA and the latter on the medium used by Garcia-Acha et al. (1966a). Preparation of lytic enzymes. The lytic enzyme complex from S. venezuela was prepared as described by Peberdy and Gibson (1971), and that from T. harzianum as described by Malard (1981) except that the culture medium was concentrated to 1/10 volume. Culture filtrate samples were lyophilised and stored desiccated at -20°C. The activity of culture filtrate samples, used as lytic preparations, was determined from their hydrolytic activity against chitine. The procedure was as follows: 2 ml of concentrated filtrate (15 mg dry weight) was incubated with 2 ml of suspension of purified chitine 0.1% (w/v) in 0.05M maleate buffer, pH 5.8, for 2 hours at 37°C. The hexosamines released by the reaction were measured by the method of Tsuji et al. (1969). In these conditions, the quantity of hexosamines was 5.3 µg/mg dry weight for Trichoderma filtrates and 26 µg/mg dry weight for Streptomyces filtrates. Buffers and stabilisers. Sucrose, mannitol, KCl and MgSO4 were used as stabilisers at different concentrations ranging from 0.4 to 1M. With the exception of MgSO4, they were dissolved in 0.1M phosphate buffer and the solution adjusted to pH 5.8. The magnesium salt was dissolved in 0.05 M maleic acid, pH 5.8. Preparation of protoplasts. PDAY Petri-dishes covered with a cellophane disc were inoculated with a mycelial suspension of 4-day-old cultures, fragmented in water with a MSE homogeniser. Cultures were incubated at 26°C for 20 hours, corresponding to the most favourable age for protoplasts production. Mycelia were collected on a 50 µm nylon mesh (which allowed removal of conidia), washed four times with the osmotic stabiliser and resuspended in stabilised buffer containing the lytic enzymes. Except when the effect of mycelial concentration on protoplast formation was investigated, all experiments were carried out with 0.1 g fresh weight mycelia suspended in 5 ml osmotic stabiliser. The mycelium was then incubated in the presence of lytic enzymes, at 26°C, with gentle shaking for 4 hours. The pre-treatment with a thiol compound was performed by incubating mycelia for 30 to 120 min. with 0.05 to 0.5% b-mercaptoethanol. After incubation, mycelia were washed four times and resuspended in 0.6 M KCl with cytohelicase alone. To test additional effects, a combination of 5 mM EDTA, pH 7, and 0.1% b-mercaptoethanol was realised. After incubation with lytic enzymes, the protoplasts were separated from the mycelial debris by filtration through a 50 µm nylon mesh. Protoplasts yields were determined by counting in a haemocytometer. Protoplast viability was monitored using methylene blue as a vital stain and protoplasts that remained colourless were considered viable (Garcia-Acha et al., 1966b). Regeneration of protoplasts. Using centri-fugation, protoplasts from the crude suspension were collected, washed twice with 0.6 M KCl and purified on a 30µ sucrose cushion as described by Bos and Slakhorst (1981). The regeneration process was investigated. A purified protoplast suspension was poured on sterile microscope slide covered with the regeneration medium (PDAY supplemented with 20% sucrose) and then incubated at 26°C. Regeneration was examined at various intervals by phase-contrast microscopy or after staining with lactic phenol blue. Regeneration frequencies were estimated by plating a known number of protoplasts on agar plates and counting the number of colonies formed after 4 days of incubation at 26°C. The regeneration medium was PDAY (1 or 2% agar) and supplemented with 20% sucrose or 0.6 M KCl. Nuclear staining. Protoplasts and mycelium were fixed for a night with Carnoy. Fixed preparations were transferred to 95, 70 and 50% alcohol solutions before hydrolyse in 5 N HCl at room temperature for 2 hours. Preparations were washed with 0.1M phosphate buffer pH 7, and then stained with Giemsa R diluted 2 and 5 fold in phosphate buffer, respectively, for mycelium and protoplasts. Enzymes and chemicals. Helicase and cytohelicase were obtained from Industrie Biologique Française, Villeneuve- la-Garenne, France.β-glucuronidase type H-5 from Helix pomatia (450.000 fishman units [FU] per gram), chitinase from Streptomyces griseus and lipase type 1 were obtained from Sigma Chemical Co., St. Louis, Missouri, USA.β-glucuronidase from H. pomatia (12 units [U] per ml) was also obtained from Merck, Darmstradt, Germany. Novozym 234 from T. harzianum was donated by Novo Industry, Bagswaerd, Denmark, while Giemsa R was purchased from Prolabo, Paris, France. Other chemicals were of reagent grade. RESULTS AND DISCUSSION Protoplasts formation. When Novozym was used as a lytic system, protoplasts began to emerge as early as 10 min. after the beginning of incubation. They were liberated from the apical and lateral zones of the hyphae, leaving empty hyphal segments. In the early stage of digestion (30 min.) small non- vacuolate protoplasts (3-6 µm) constituted most of the population (Fig. 1). Continuous incubation resulted in the liberation of large vacuolate protoplasts. Thus, after 4 hours of incubation, the population of protoplats was heterogeneous in size, varying from 3 to 15 µm (Fig. 1). This was observed with the three lytic system tested : Novozum, b-glucuronidase, and cytohelicase. The small non-vacuolate protoplasts were probably released from hyphal tips, while the larger vacuolate from more distal regions as previouly observed on Aspergillus sp. by Gibson and Peberdy (1972) and Thomas et al. (1984). Protoplasts were also heterogeneous in their nulear material. Nuclear staining showed that only 60% of these protoplasts were nucleated (Fig. 2). This result is different from that obtained by Garcia-Acha et al. (1966b) with Fusarium culmorum in which most of the protoplasts were nucleated. In our case, although the number of nuclei between two septa was most often between 1 and 4 (Fig. 2), a great number (40%) of protoplasts were nucleated. This may be explained by the distribution of the hyphal contents into several protoplasts coming out of the same compartment (Villanueva, 1966). We also observed that the size of protoplasts was not strictly related to the number of nuclei since protoplasts of 12 µm were either anucleate or contained 1 to 4 nuclei. Viability of freshly isolated protoplasts assessed by the methylene blue staining technique was repeatedly above 85%. Viability was not affected by storage at 2°C for 24 hr. prolonged storage at 26°C induced protoplast swelling up to a 30 µm diameter. Lytic activities of various enzyme pre-parations.To find a suitable lytic system, different enzyme preparations were tested for their lytic activity against F. solani mycelium. We tried our own lytic complexes from T. harzianum and S. venezuelae and different commercial preparations: snail digestive juices including helicase, cytohelicase and b-glucuronidase and Novozym from T. harzianum. The yields of protoplasts obtained with the optimal concentrations of the various lytic enzymes are shown in Table 1. Novozym and b-glucuronidase (sigma) were the most effective in promoting the release, respectively, of 109 and 3.108 protolpasts g-1 fresh weight. Other enzyme preparations gave yields 50 to 200 times lower. In addition, the synergistic action of lytic enzymes on protoplast release was tested by combinating helicase either with chitinase, lipase or Streptomyces filtrate. As shown in Table 1, the association with chitnase did not stimulate the production of protoplasts as reported in other situations (Thomas et al., 1979; Hamlyn et al., 1981; Picataggio et al., 1983). Moreover, a decrease in protoplasts production was observed as the chitinase concentration increased. On the contrary, the helicase-lipase combination resulted in enhanced protoplast yield as noted by Garcia-Acha et al. (1966c) and Coudray and Canevascini (1980). A similar enhancement was noted for the helicase-Streptomyces filtrate combination. However, the yields obtained with these two combinations remained clearly lower than that obtained with Novozym alone. In spite of its weaker efficiency, further experiments were carried out with cytohelicase because it was more readily available than the more efficient enzymes.
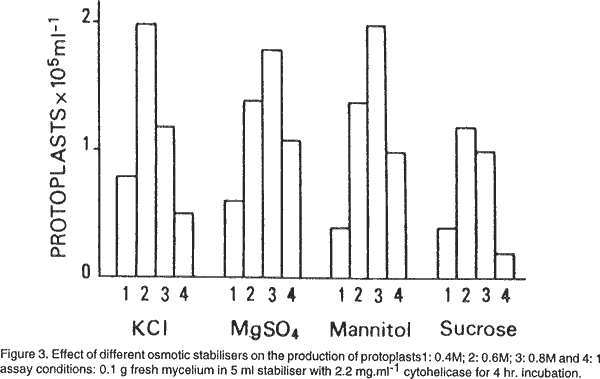
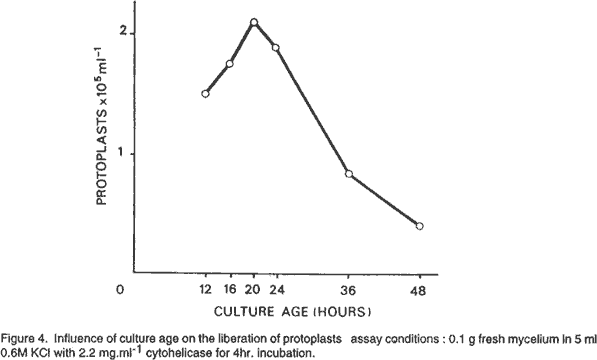
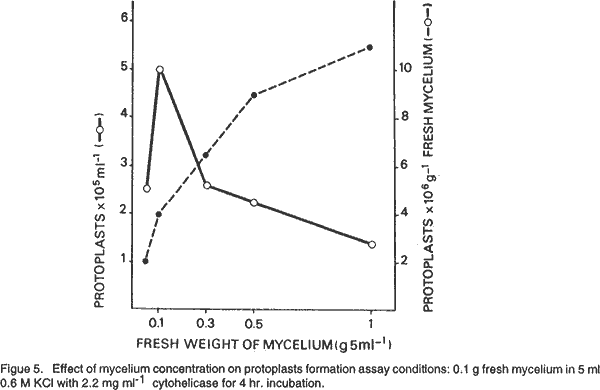
Osmotic stabilisers. We used as osmotic stabilisers, minerals salts and sugars. As shown in Figure 3 KCl, MgSO4 and mannitol were more effective than sucrose. The highest yields were obtained with 0.6 M KCl and 0.8 M mannitol. Protoplasts liberated with 0.8 M MgSO4 were largely vacuolate as reported by Peberdy (1979). Age of mycelium. Mycelia from cultures of different ages, from 12 to 48 hours old, adjusted to equivalent concentrations were tested for protoplast production. Maximal yields were obtained from 20 hr. old mycelium (Fig. 4). Older cultures resulted in a marked decrease in protoplast yield. Effect of mycelium concentration in the lytic digestion. Mycelium from a 20 hr. old culture, adjusted to concentration ranging from 0.1 to 1 g fresh weight was incubated with cytohelicase. The number of protoplasts per ml increased with increasing mycelium concentrations up to the limiting enzyme concentration (Fig. 5). However, the maximal yield (protoplasts g-1 fresh mycelium) was obtained for the ratio 0.1 g/11 mg cytohelicase. The protoplast concentration can be increased by treating more mycelium with a quantity of enzyme in the same ratio than above, the yield remaining unchanged. Pre-incubation of mycelium with a thiol compound. Microscopic observations showed that many hyphae remained intact after 4 hr. incubation with cytohelicase indicating that the enzyme was not totally effective in digesting the cell wall. Thus, the effect of mycelium preincubation prior to digestion with a thiol compound known to exhibit a stimulating effect on protoplast release (Peberdy, 1979; Harris, 1982) was investigated. Mycelium was pre-treated for 1 hour with 0.1% (v/v)β-mercaptoethanol. This condition was the most effective and resulted in a 2-fold increase in the number of protoplasts. Addition of 5 mM EDTA did not modify the yield of protoplasts. Regeneration of protoplasts. The first visible signs of the regeneration process could be seen after 4 hr. incubation on the regeneration medium. Some protoplasts increased in size, some lost their spherical shape, while others emitted a bud which remained attached to the protoplast. After 8 hr. regeneration, many cellular aggregates were observed and after 10 hr., the first hyphal tubes emerged. At least three different patterns of regeneration were observed. In the first, protoplasts gave yeast-like bodies grouped in a chain of 3 to 15 cells. Hyphae emerged most often from the terminal cells of a chain. In the second, protoplasts directly produced one germ tube. The third type occurred through a complicated process involving the formation of polymorphic structures from which hyphal tubes arised. The regeneration patterns were similar, whatever the lytic enzyme used for the protoplast release. However, the regeneration frequency may differ. Under optimal conditions for reversion we established that 3 to 30% of protoplasts obtained with cytohelicase were able to regenerate while 20 to30% regenerated with Novozym. REFERENCES
The following images related to this document are available:Line drawing images[cs01016e.gif] [cs01016c.gif] [cs01016b.gif] [cs01016d.gif] [cs01016a.gif] | ||||||||||||||||||||||||||||
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}