 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
African Crop Science Journal, Vol. 19, No. 4, 2011, pp. 357-367 Characterisation of bacterial brown spot pathogen from dry bean production areas of South Africa H.T.H. Muedi, D. Fourie and N.W. Mclaren1
Agricultural Research Council - Grain Crops Institute, 114 Chris Hani Street, Potchefstroom,
2520, South Africa
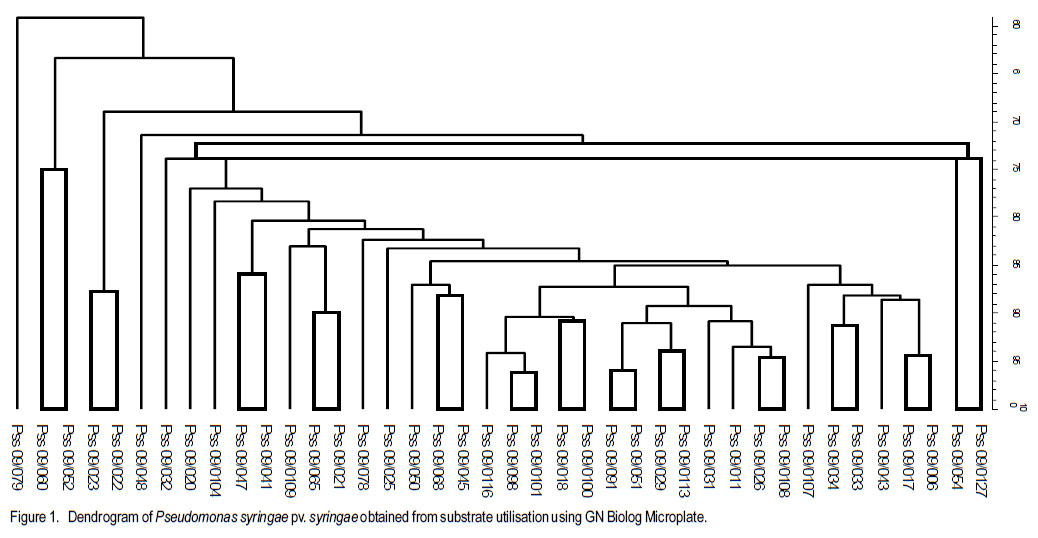
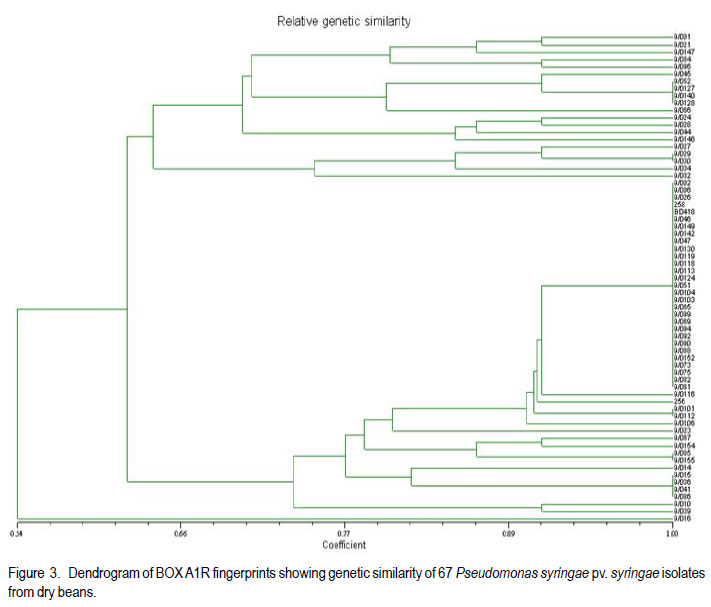
Code Number: cs11033 ABSTRACT Pseudomonas syringae pv. syringae (Pss) causes bacterial brown spot (BBS) of beans (Phaseolus vulgaris L.), with yield losses of up to 55% in South Africa. Pss has a wide host range and for many of these, the pathogen has been biochemically and genetically characterised. However, few studies have been conducted on Pss isolated from beans. The aim of this study was to assess the biochemical and genetic variability of Pss isolates collected from dry bean producing areas in South Africa. Pure isolates were subjected to LOPAT tests and SyrB gene assessment. Biolog GN Microplates were used to assess carbon substrate utilisation. The SyrB gene was present in 42% of isolates. The Biolog GN Microplates showed biochemical variation among isolates. Variable genomic patterns were observed in 48.5% by the BOX A1R primer and in 37.1% of isolates by the ERIC 2 primer. Thus, variability exists in Pss populations of dry beans. Key Words: Phaseolus vulgaris, Pseudomonas syringae, SyrB gene RÉSUMÉ Pseudomonas syringae pv. syringae (Pss) cause de la maladie de tâche bactérinne brune (BBS) de haricot (Phaseolus vulgaris L.), et des pertes de rendement allant jusqu’à 55% en Afrique du Sud. Pss possède une large gamme d’hôtes et pour la plupart d’entr’eux, le pathogène a été biochimiquement et génétiquement caractérisé. Cependant, peu d’études ont été conduites sur le Pss isolé du haricot. L’objet de cette étude était d’évaluer la variabilité biochimique et génétique des isolats de Pss collectés des milieux producteurs de haricot sec en Afrique du Sud. Des isolats pures étaient soumis aux tests LOPAT et évaluation du gène SyrB. Les microplats Biolog GN étaient utilisés pour évaluer l’utilisation du substrat de carbone. Le gène SyrB était présent dans 42% des isolats. Les microplats Biolog GN ont montré une variation biochemique parmi les isolats. De modèles génomiques variables étaient observés dans 48.5% par le BOX A1R primer et dans 37.1% des isolats par le ERIC 2 primer. Ainsi, il exite une variabilité dans les populations de Pss de haricot sec. Mots Clés: Phaseolus vulgaris, Pseudomonas syringae, gène SyrB INTRODUCTION Dry bean (Phaseolus vulgaris L.) is an important staple food and cash crop for subsistence and commercial farmers of South Africa (National Department of Agriculture, 2003). In frost-free areas, production by subsistence farmers is done twice in a year. Dry bean is at present regarded as one of the most important field crops in South Africa on account of its high protein content (2224%) and dietary benefits (Liebenberg et al., 2002). Although dry seed is the desired product by consumers, production is hampered by a number of factors including bacterial diseases. Pseudomonas syringae pv. syringae (Pss) has a wide host range, broader than other pathovars both as epiphyte and pathogen, causing disease in more than 180 species of plants in several unrelated genera (Hirano and Upper, 1990). Pseudomonas syringae pv. syringae is an important pathogen of beans world wide, that causes bacterial brown spot (BBS) with associated yield losses of up to 55% in South Africa (Serfontein, 1994). Bacterial brown spot was reportedly the most widespread bacterial disease of dry bean in South Africa, occurring in 93% of seed production fields and in 100% of commercial fields (Fourie, 2002). All commercially grown South African dry bean cultivars are susceptible to BBS. The plant toxin syringomycin, a cyclic lipodepsinonapeptides metabolite, is responsible for damage to bean plants (Quegley and Gross, 1994). Syringomycin, among other toxins, is produced and conserved among Pss strains possessing the SyrB gene (Quegley and Gross, 1994; Zhang et al., 1995; Arrebola, 2009). Syringomycin is one of the major virulence factors of Pss, and genes responsible for their production are found in Pss and not in other related pathovars (Mo and Gross, 1991; Quigley and Gross, 1994). The primers used to detect the SyrB gene amplify the 752-bp band in Pss strains (Natalini et al., 2006). The amplification of the SyrB gene is, therefore, a strong identification technique for Pss isolates or strains. Relationships among Pss isolated from many other host plants have been studied through biochemical (substrate utilisation) and molecular analysis (Little et al., 1998; Natalini et al., 2006; Vicente and Roberts, 2007). Availability of biochemical diagnostic kits has made identification of Pss easier and much more reliable than conventional methods (Khezri et al., 2010). Biochemical assessments alone are reportedly not reliable for differentiating strains at or below pathovar level (Roos and Hattingh, 1987). The importance of repetitive sequence PCR fingerprints with BOX and enterobacterial repetitive intergenic consensus (ERIC) primers, which are short repetitive DNA sequences, has been widely reported (Little et al., 1998; Scortichini et al., 2003; Vicente and Roberts, 2007). Universal rep-PCR primers that generate highly reproducible, strain-specific fingerprints that can differentiate bacterial strains at species and sub-species levels, have been designed from these DNA sequences (Louws et al., 1994). The relationship between Pss strains infecting dry bean plants in South Africa is currently unknown. The objective of this study was to assess the biochemical and genetic variability of the Pss isolates from the major dry bean producing areas of South Africa. MATERIALS AND METHODS Isolation of Pss. One hundred and forty isolates were collected from 24 locations in five different provinces of South Africa [KwaZulu Natal (16 isolates), Free State (56 isolates), Mpumalanga (33 isolates), Limpopo (22 isolates) and North West (13 Isolates)] during the 2008/2009 growing season. Leaves and pods with typical bacterial brown spot symptoms were collected during pod stage from 6 home gardens, 13 National Cultivar Trials, 8 subsistence and 14 commercial farms. Leaves were stored in brown paper bags during transportation to the laboratory. Leaves were rinsed under running tap-water for 10 min, surface-sterilised for 3 min in 3.5% sodium hypochlorite, and rinsed twice in sterile water for 1 min each. Leaves were macerated in a droplet of sterile water and the macerate was streaked onto King’s B medium (King et al., 1954) prior to incubation at 26 oC for 72 hr. LOPAT, carbon source utilisation and pathogenicity tests. Fluorescent colonies, typical of Pseudomonas spp., were selected under UV-light and purified on King’s B medium. Isolates were tested for oxidase reaction (-) and levan production (+); and mannitol, sorbitol and inositol carbon sources were used to distinguish Pss from P. savastanoi pv. phaseolicola isolates. Serological identification was performed using a commercial Pss-specific agglutination antibody (Express Kit, NEOGEN Europe Ltd., Scotland, UK). Approximately 10 µl of test reagent was placed on the testing card and mixed with test bacterial colonies using sterile toothpicks. Positive reaction was observed as granular agglutination, while transparent (no agglutination) demonstrated negative reaction. Positive and negative controls of the Express Kit were included. (i). Potato rot test. Fresh potato tubers were washed, alcohol flamed, peeled and sliced into approximately 7 mm width. The slices were placed in 90 mm diameter petri-dishes and sterile distilled water was added to a depth of half the slice. One hundred micro-litres of a 24 hr old bacterial suspension in nutrient broth was pipetted into a 3 mm diameter well on the centre of each slice. Positive results were indicated by lack of rotting, while rotting suggested negative results. Erwinia carotovora (Pectobacterium carotovorum) was used as negative control and un-inoculated nutrient broth as positive control (Ignjatov et al., 2007). (ii). Hypersensitive test. To distinguish between plant-pathogenic and non-pathogenic isolates, tobacco (Nicotiana tabacum) hypersensitivity tests were conducted according to the method of Klement (1963). Each isolate was suspended in sterile distilled water (1x106 cfu.ml-1), determined by serial dilution, and inoculated on the underside of fully expanded tobacco leaves using a syringe. Sterile distilled water was used as negative control. Plants were kept under greenhouse conditions (26 °C during daytime and 18 °C during night time) and observations for a hypersensitive reaction were conducted after 24 hr. (iii). Bean pathogenicity. The 1x106 cfu ml-1 suspension was also used to spray to wetness the underside of leaves of a susceptible cultivar, Sederberg, at two-leaf stage using a DeVilbiss atomiser spray. Inoculated plants were placed in a dew chamber for 48 hr. Negative control plants were inoculated with sterilised distilled water. Plants were evaluated for disease reaction at 5-7 days after inoculation with negative (-) and positive (+) assigned for absence and presence of symptoms, respectively. Isolates were also inoculated onto detached dry bean pods using a hypodermic needle as described by Cheng et al. (1989). Sterile distilled water was used as negative control. Isolates that completely lacked the characteristics of Pss were discarded, hence they were scaled down to 132. Carbon substrate utilisation and biochemical test. A total of 132 isolates were characterised by means of sole carbon source utilisation profiles using the Biolog GN Microplates (Biolog, Hayward, CA) according to the manufacturers’ instructions. Twenty four hour old isolates grown on King’s B medium were homogenously suspended in sterile distilled water before inoculation on the profiles. Results of the Biolog GN Microplates were recorded after 24- and 48hr incubation at 26 oC. Biochemical reactions were visually observed and subjected to a microtiter plate reader (Bio-Tek ELx 800) at 620 nm absorbance level. Data, were entered into the Bionumerics software (version 4.5) and subjected to cluster analysis using the unweighted pairgroup-average method (UPGMA). The identity of isolates were further confirmed using the Analytical Profile Index, API 20E (bioMerieux, Marcy l’Etoile, France), according to the manufacturers’ instructions. Homogeneous suspensions (1x106) were inoculated in the tubes of API 20E kits. Data were recorded after 24 hr incubation at 26 oC and seven-digit profiles developed for every isolate. Profiles were identified using the APILAB version 4.1 identification program (BioMérieux). GENETIC DIVERSITY OF ISOLATES DNA extraction. Genomic DNA was extracted from fresh bacterial isolates cultured on King’s B medium and incubated at 26 oC overnight using PrepMan Ultra Sample Preparation Reagent (PUSPR) method, (Applied Biosystems, South Africa). A loopful of bacterial cells was transferred into 1.5 ml sterile tubes and suspended in PUSPR. Cells were pelleted by centrifugation at 12,000 rpm for 10 min and pellets resuspended in sterilised distilled water. Precipitated DNA was transferred into new sterilised, properly labelled 1 ml tubes. DNA was diluted to 30 ng ml-1 and stored at -80 oC. Production of cyclic lipodepsinonapeptides. PCR amplification was conducted in a 0.5 ml microtube using a MBS satellite 0.2 G thermal cycler. The 21-mer oligonucleotides primers, B1 (5’-CTT TCC GTG GTC TTG ATG AGG-3’) and B2 (5’-TCG ATT TTG CCG TGATAG TC-3’) (Whitehead Scientific, South Africa), as suggested by Sorensen et al. (1998), were used for the detection of syrB gene coding for the production of cyclic lipodepsinonapeptides (e.g. syringomycin) in Pss strains. The 20 ml PCR reaction contained 1X PCR buffer (50 mM KCl, 10 mM Tris-HCl [pH 8.3]), 0.5 mM concentration of each primer, a 200 mM concentration of each dNTP, 1.5 mM of MgCl2, 0.025 U of Taq polymerase enzyme per ml (Promega Corp., Madison, WI), 200 ng of genomic DNA. The PCR conditions of Sorensen et al. (1998) were used. Amplified fragments were separated in 1.5 % agarose gel stained with ethidium bromide (10 mg ml-1). The 1xUNTAN buffer was used and the gel photographed using documentation system (Bio-Rad Gel Doc 1000). Bands were scored as either present or absent. Repetitive PCR. The rep-PCR products of the primers BOX A1R (5’-CTA CGG CAA GGC GAC GCT GAC G-3’) and ERIC 2 (5’-AAG TAA GTG ACT GGG GTG AGC G-3’) obtained from Fermentas, Inqaba Biotec, South Africa, were used to fingerprint isolates following PCR conditions of Renick et al. (2008). Amplified fragments were separated in 1.5% agarose gel, stained in ethidium bromide (10 mg ml-1) for 30 min, destained for 15 min in ultra pure water and visualised under the Bio Rad XR Gel Documentation System, and scored. Bands were scored irrespective of their intensity. A 200-bp O’RangeRuler molecular weight marker was used (Fermetas Life Sciences, South Africa). Experiments of each primer were repeated at least four times to confirm the reproducibility of banding patterns. The rep-PCR banding patterns were compared to Pss banding patterns of Little et al. (1998) and Scortichini et al. (2003) from other host plants. The scoring of the banding patterns of the rep-PCR was conducted using the NTSYS software version 2.11 S and the cluster analysis was developed using the UPGMA method. RESULTS LOPAT and pathogenicity tests. All fluorescent isolates tested were levan-positive, oxidasenegative, potato soft rot-negative, arginine dihydrolase-negative and tobacco hypersensitivity-positive (LOPAT test group Ia) (Data not shown). Variable reactions were recorded on susceptible cultivar Sederberg where the majority of isolates caused severe BBS disease. Isolates that were virulent on Sederberg were generally aggressive in the detached pod test. The diameter of water-soaked lesions on detached green bean pods varied from 1-5 mm, while no water-soaked lesions were observed in control treatments (Data not shown). Carbon substrate utilisation and biochemical test. The Biolog GN Microplates identified 30% of isolates assessed as pure Pss (Fig. 1). Carbon source utilisation patterns were more pronounced at 48- than 24-hr incubation. Genetic diversity of isolates. The 752-bp fragment of the SyrB gene was present in 55 of the 132 isolates (Fig. 2.), where 32 of the 55 were aggressive in pathogenicity tests. The 752-bp fragment was absent in the majority of isolates that tested negative for pathogenicity. The 752bp fragment did not amplify in all isolates that were identified as Pss through biochemical and physiological evaluations. Variation in genomic patterns was observed in 48.5% by the BOX A1R primer (Fig. 3) and in 37.1% by the ERIC 2 primer (Fig. 4) of aggressive and non-aggressive isolates combined. A few isolates amplified in one primer and not in the other. While some isolates did not give any reproducible genomic rep-PCR profiles, BOX A1R and ERIC 2 primers yielded scores of 11 and 15, respectively, clearly resolved bands for the UPGMA analysis. The fragments ranged in size from 400- to 3000-bp for BOX A1R and from 500to about 3600-bp for ERIC 2 primers. Isolates in group A of the BOX A1R primer appear to have genomic fingerprint of 60% similarity with those in B, however, majority of isolates in group A possessed the syrB gene (Fig. 2 and Fig. 3). Approximately 65% of genetic similarity was observed between group A and B of the ERIC 2 primer. Isolates belonging to group C for both rep-PCR primers lacked the syrB gene and were not pathogenic on susceptible cultivar Sederberg. To a large extent, ERIC 2 primer grouped isolates according to areas of collection (Fig. 4), it was, however, not the case with BOX A1R primer. Isolate BD 418 from wild cucumber (Cucumis myriocarpus) was clustered in group B of the BOX A1R primer together with the majority of isolates from dry bean plants. DISCUSSION Majority of isolates caused typical BBS symptoms on susceptible cultivar Sederberg and on detached bean pods pathogenicity tests (Data not shown). The isolates possessing the SyrB gene were especially highly aggressive on susceptible cultivar Sederberg, and that must have been due to the phytotoxin syringomycin. The SyrB gene controls the production and conservation of syringomycin (Quegley and Gross, 1994; Zhang et al., 1995). Scortichini et al. (2003) and Natalini et al. (2006) also observed strong aggression from Pss strains (from other host plants) possessing the SyrB gene. Forty two percent of isolates possessed the SyrB gene, which controls the production and conservation of the phytotoxin syringomycin (Fig. 2). Khezri et al. (2010) reported variation in syringomycin production among Pss strains from other hosts such as sugarcane, apricot, peach, almond, sweet cherry, wheat, barley, bellis and hibiscus. The production of syringomycin by Pss strains from diverse range of host plants, including bean, causes necrosis of leaves, pods and stems by inducing pore formation and disrupting plant cell membrane (Gross et al., 1984; Iacobellis et al., 1992). These toxins are secondary metabolites of diverse chemical structures and are effective at very low concentrations (Mitchell, 1991). Syringomycin has been used as a determinative characteristic in identifying pathogenic strains of Pss (Schaad et al., 1989; Young, 1991). The synthesis and export of syringomycin, which is one of the major virulence factors, is genetically controlled and genetic variability was observed when strains were subjected to different cultural conditions (Mo and Gross, 1991). The SyrB gene responsible for this action is found in Pss and not in other related pathovars and also revealed considerable diversity among Pss strains tested (Quigley and Gross, 1994). This study revealed that some isolates lacking the SyrB gene are virulent in pathogenicity test, while other isolates possessing the gene do not induce symptoms (Fig. 2). Little et al. (1998) indicated that all of the Pss strains tested in their study had the genetic potential to produce syringomycin. This suggests that all isolates assessed in the present study and were found to be possessing the SyrB gene, but failed to induce symptoms Sederberg, might still have the potential to produce syringomycin. However, Zeller et al. (1997) indicated that not all Pss strains produce syringomycin, thus agreeing with findings of this study. Biochemical variability among Pss isolates was observed through carbon-source utilisation. Availability of biochemical diagnostic kits has made identification of Pss easier and much more reliable than conventional methods (Khezri et al., 2010), and has been used extensively (Chase et al., 1992). In this study, 30% of isolates were successfully identified as Pss; however, some were classified as different pathovars of no interest to this study. This low percentage could be in line with observations by Roos and Hattingh (1987) that biochemical assessments alone are not reliable for differentiating strains at or below pathovar level. However, Jones et al. (1993) reported that Pss was distinctively identified from other P. syringae pathovars using the carbon substrate utilisation method. Carbon source utilisation patterns were more pronounced at 48than 24-hr incubation and this could be due to increased bacterial populations at 48 hr. The API 20E profiles demonstrated that 81% of isolates tested were Pss. Putative Pss isolates were further subjected to molecular assessment. Güven et al. (2004) reported that 55 P. syringae pv. phaseolicola strains differentially utilised carbon sources of the Biolog GN2 Microplate. Through similarity coefficients, this system revealed that Pss isolates in this study are closely related; a finding that concurs with that of Güven et al. (2004) who observed close relationships of P. syringae pv. phaseolicola strains (group representatives: D34, F14, 2215, 234.1 and 52). The API 20E kits identified 81% of isolates as Pss in this study. Although O’hara et al. (1992) indicated that API 20E is approximately 88.494.6% accurate at 24 hr incubation and could be higher at 48 hr, biochemical reactions in this study were satisfactory at 24 hr incubation and no improvement was observed at 48 hr. In addition, O’hara et al. (1992) reported that API 20E profiles are reliable for identification of enterics. With the exception of two isolates as indicated by BOX A1R and ERIC 2 primers, rep-PCR profiles show close relationships among a larger group of Pss isolates tested (Fig. 1 and Fig. 2). The fingerprints profiles revealed that genetic diversity occurs even though there was no definite distinction among isolates from different farming or experimental systems. This could suggest that Pss occurred in South Africa long before the first epidemics were reported (Serfontein, 1994) only in low densities or as epiphytes. Legard et al. (1993) indicated that diversity within Pss exists, supporting the findings of this study. ERIC 2 primer showed close genetic relationships among majority of isolates collected from the same dry bean production area in this study. Mazzaglia et al. (2011) reported varying results on P. syringae pv. actinidiae where some of the strains were grouped according to geographical areas of collection and others not. Strains of Pss that are adapted to a specialised niche might be the result of a genetic isolation, resulting in a genetically homogenous population of Pss (Little et al. 1998; Khezri et al., 2010). Banding patterns differed between BOX A1R and ERIC 2 profiles, and also did not match patterns shown by Natalini et al. (2006) and Scortichini et al. (2003) using Pss isolates from other host plants. From the present study, there was no genetic distinction between isolates from dry beans and BD 418 from C. myriocarpus. However, in a study by Legard et al. (1993), Pss strains from beans formed a separate cluster within the pathovar when compared with strains from other host plants. Little et al. (1998) indicated that additional studies by other genetic characterisation methods support the hypothesis that variation was greater among strains from pathovars with wide host ranges, such as Pss. Other pathovars with a more restricted host range, such as P. syringae pv.morsprunorum and -tomato, had low or no diversity in their ERIC profiles (Little et al., 1998). Scortichini et al. (2003) indicated that diversity was observed among isolates from the same host plant as well as isolates from the same site, isolated at the same time. The rep-PCR has been shown to be a quick and reliable method to differentiate between plant-pathogenic bacteria at or below pathovar level with highly reproducible results (Louws et al., 1994). It was reported that Pss represents a good example of genetic and pathogenic variability (Scortichini et al., 2003). Biochemical tests and molecular analyses yielded reliable results when applied successively on similar isolates. Findings of this study conclude that significant biochemical and genetic diversity exists among Pss isolates from dry bean producing areas of South Africa. Pseudomonas syringae pv. syringae isolates possessing the SyrB gene are more aggressive on susceptible dry bean host plants than those lacking the gene. ACKNOWLEDGEMENT The Biotechnology staff of the ARC-Grain Crops Institute, South Africa, provided supportive expertise. The Agricultural Research Council (ARC) provided facilities. The Dry Bean Producers Organisation and the Department of Science and Technology funded this research. Partial support received from the Pan-Africa Bean Research Alliance (PABRA) is gratefully acknowledged. REFERENCES
Copyright © 2011, African Crop Science Society The following images related to this document are available:Photo images[cs11033f1.jpg] [cs11033f2.jpg] [cs11033f3.jpg] [cs11033f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}