 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Dermatology, Venereology & Leprology, Vol. 69, No. 3, May-June, 2003, pp. 229-231 Case Report Cornelia de Lange syndrome K. Muhammed, B. Safia Department of Dermatology and Venereology, Medical College, Kozhikode-673008,
India.
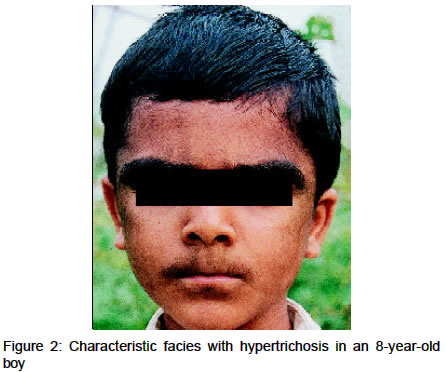
Abstract Two cases of Cornelia de Lange syndrome with similar phenotypic features are reported. Key Words: Cornelia de Lange syndrome, Dwarfism Introduction Cornelia de Lange syndrome (CDLS), also known as Brachman de Lange syndrome or Amsterdam dwarf, is characterized by a typical facies along with hypertrichosis, cutis marmorata and a bluish discoloration of the facial skin. Skeletal abnormalitites, mental retardation and abnormal cry are also present. Credit for the description of the syndrome is generally given to Cornelia de Lange, a Dutch pediatrician who reported it in 1933, almost two decades after Brachman's original description. The incidence is 1 in 10,000 live births. Early death usually occurs in these patients. Although several significant cutaneous manifestations are seen in CDLS, the dermatological literature has little information about them. The characteristic facies includes a bluish cast about the eyes, nose and mouth, overgrowth and confluence of eyebrows (synophrys), long curly eyelashes, a small red nose with low bridge, anteverted nostrils, increased inter-alar distance and a broad upper lip, which is thin and down-turned, producing a grim mask-like appearance. The ears may be low set. Hyperplastic epidermal ridges of the palms, soles, fingers and toes may occur. There is a possible correlation between epidermal ridge pattern and survival,1 in that the syndrome appears to be less severe in patients with a relatively normal ridge pattern. An important feature of the syndrome is the striking delay in the maturation of structure and function of most organ systems. A dominant mutation has been postulated as the most likely etiologic possibility. Case Report Case 1 An 8-year-old boy, born of a non-consanguinious marriage,
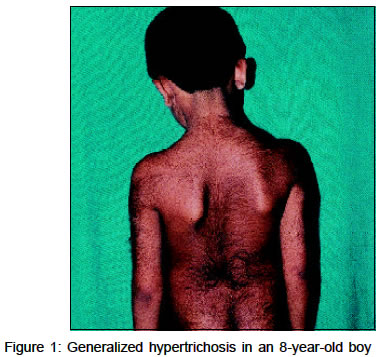
presented with generalized hypertrichosis since early childhood (Figure
1).
There was a history of delayed milestones, and poor performance at school.
His birth weight had been 2.8 kg. The present height, weight and head circumference
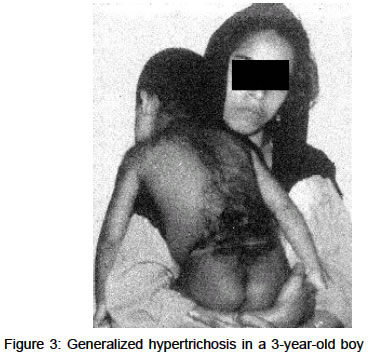
were 125 cm, 21 kg and 53.3 cm respectively. The patient had a low The baseline blood and urine investigations and thyroid function tests were within normal limits. Serum FSH and testosterone levels were lower than normal. An X-ray of both hands showed decreased bone age. Ultrasonography of the abdomen did not reveal any organomegaly. The child had multiple retained deciduous teeth and showed missing permanent teeth on X-ray. Shaving of the facial hair was the only treatment being followed. Laser hair removal and extraction of deciduous teeth with construction of prosthesis were suggested. Case 2A 3-year-old boy was brought to our outpatient department with generalized hypertrichosis since birth, and mental retardation. He was the only child of a non-consanguinious marriage, born of a full term normal delivery with a birth weight of 2.9 kg. There was a history of posterior urethral valve correction at the age of one year. Delayed developmental milestones were present. His height,
weight and head circumference were 88 cm, 10 kg and 44.5 cm respectively, with
the expected normal values being 100 cm, 14 kg and 48 cm respectively at this
age. He had hypognathism, clinodactyly and a characteristic mask Discussion Cornelia de Large syndrome is so characteristic that once a case has been seen, another may be identified immediately because of the strikingly similar phenotype. The cry alone suffices to suggest the diagnosis without having seen the patient. Our patients had all the characteristic facial features, generalized hypertrichosis and decreased skeletal and mental growth. Undescended testes are reported in 73% of male patients. Patients may have decreased gamma globulin and increased serum alpha ketoglutarate and serum glutamate levels, which may help in the diagnosis. Postmortem examination may show developmental retardation of all major organs of the body, with the exception of the liver.2 The etiology and recurrence risk of CDLS are unknown. The syndrome may be the result of an inherited metabolic error.3 No environmental cause has been discovered. Most cases are sporadic, although an autosomal dominant inheritance has been suggested. CDLS without severe mental retardation has been reported by Pashayan and co-workers.4 Duplication corresponding to bands q25- q29 of chromosome 3 has been reported.5 Temporary or permanent removal of the hair is the only treatment that can be offered, for cosmetic improvement. References
Copyright 2003 - Indian Journal of Dermatology, Venereology & Leprology. Free full text also available from: http://www.ijdvl.com The following images related to this document are available:Photo images[dv03012f3.jpg] [dv03012f1.jpg] [dv03012f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}