 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
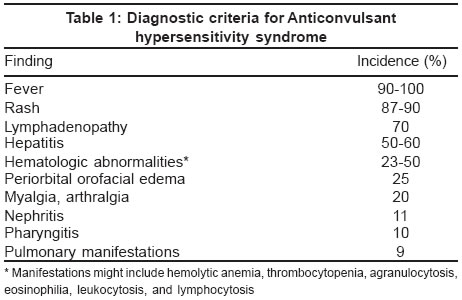
Indian Journal of Dermatology, Venereology, Leprology, Vol. 70, No. 1, January-February, 2004, pp. 48-51 Residents' Page Pseudolymphoma syndrome revisited Goel Ashima , Walia RLS Department of Dermatovenereology, Rajindra Hospital, Patiala Code Number: dv04016 INTRODUCTION Anticonvulsants have long been recognized as a cause of hypersensitivity reactions. Phenytoin hypersensitivity can be traced to 1916, when phenylethylhydantoin (phenytoin sodium), which was used to treat children with Sydenham chorea, was found to induce a hypersensitivity reaction. Phenytoin was then known as a "nerve sedative," and the hypersensitivity reaction, "nirvanol sickness," resolved on discontinuation of the hydantoin.[1] The constellation of systemic manifestations was first described as the Dilantin sensitivity syndrome in 1950.[2] The syndrome was referred to as anticonvulsant hypersensitivity syndrome (AHS) in 1988, after its occurrence was found to be related to antiepileptic drugs other than phenytoin.[3] Antiepileptic drugs have been implicated as etiologic agents for a variety of dermatologic eruptions, including allergic or leukocytoclastic vasculitis, Stevens-Johnson syndrome (erythema multiforme major), and toxic epidermal necrolysis.[4],[5] These dermatoses exhibit clear diagnostic features that differentiate them from AHS. Pathophysiology The mechanism by which the antiepileptic drugs induce AHS is not well understood. Most data have been obtained for the antiepileptic drugs whose chemical structure is based on an aromatic ring (carbamazepine, phenobarbital, phenytoin, and primidone). Oxidation renders these compounds to nontoxic hydroxylated metabolites. The arene-oxide intermediate of this reaction might be responsible for toxic interactions with the cytochrome P-450 system.[3],[6],[7],[8] The cytochrome P-450 mixed-function oxidase system has been implicated in many drug reactions and interactions. Carbamazepine, phenobarbital, and phenytoin induce cytochrome P-450 3A (CYP3A), a subfamily of the cytochrome P-450 system.[9],[10] Using rat liver microsome-derived cytochrome P-450, it has been shown that the serum of antiepileptic drug-exposed patients contains predominantly anti-rat CYP3A antibodies. The amino acid sequence of rat CYP3A differs from homologous human CYP3A by only one amino acid. Site-directed mutagenesis in vitro has confirmed that alteration of the rat cytochrome sequence to that of the human cytochrome sequence renders the rat cytochrome nonantigenic. Arene-oxide metabolites of antiepileptic drugs might alter these homologues to more closely resemble rat CYP3A, initiating an autoimmune attack on the target organs where these cytochromes are produced: stomach, liver, intestine, and lung. Predisposed patients might be unable to detoxify these metabolites adequately.[3],[9],[11],[12],[13] It has also been proposed that AHS is virally mediated in association with human herpesvirus 6 in a fashion similar to the viral association of Epstein-Barr virus and ampicillin or the proposed association of sulfa hypersensitivity in HIV disease. Each of these three viral associations invokes an immunocompromised state that effectively disables detoxification pathways.[12] Other investigators have shown in vitro and in vivo evidence that carbamazepine and phenytoin mimic viral infection by activating CD4+ and CD8+ T cells, with the concomitant production of interleukin 5, the main maturation factor for eosinophils. These findings are similar to other superantigenic processes, eg, toxic epidermal necrolysis,[14] and contribute to our understanding of the exanthems associated with AHS. Lamotrigine, a new antiepileptic drug, has been implicated with the cutaneous manifestations of Stevens-Johnson syndrome and toxic epidermal necrolysis almost always when used in combination with valproic acid.[15] Diagnosis The current diagnostic criteria are listed in [Table - 1].[3],[16] Fever can precede other manifestations by several days and can be low-grade, but it is generally high, spiking, 100°F to 104°F (38°C to 40°C). The rash occurs 3 weeks to 3 months after the antiepileptic drug is initiated and may be pruritic.[5],[8] Clinical cases of AHS have the classic morbilliform or erythrodermal exanthem, with subsequent desquamation. Lymphadenopathy can be local or generalized. Hepatic involvement ranges from a transient rise in transaminases to liver necrosis with fulminant failure. Coombs-negative hemolytic anemia, thrombocytopenia, agranulocytosis, eosinophilia, or lymphocytosis might be observed.[5],[6] Profound leukocytosis can manifest with white blood cell counts previously reported as high as 43 x 103/µL.[16] Pulmonary findings are less common but can manifest with such serious outcomes as bronchiolitis obliterans with organizing pneumonia.[17] The most disturbing hematologic finding is that of atypical lymphocytes consistent with lymphomatous transformation. In 1959, histologic lymph node findings among all 7 patients with AHS were consistent with a lymphoma-like pattern.[18] This finding is now commonly referred to as "pseudolymphoma," "pseudo-Sézary syndrome," or "pseudolymphoma syndrome," consistent with the findings of cytologic or histologic lymphoma-like cells.[18],[19] Some investigators suggest that there are actually two separate hypersensitivity syndromes mimicking lymphoma. The first is the hypersensitivity syndrome described above; the second is more insidious, the initial manifestation being cutaneous nodule and plaque formation months after an antiepileptic drug is initiated. These latter cutaneous manifestations reveal histologic pseudolymphoma, which resolves with discontinuation of the antiepileptic drug but increases the likelihood of lymphomatous transformation later in life. These investigators propose a formal name of "drug rash with eosinophilia and systemic symptoms" for the former condition, currently known as AHS.[18] Perhaps AHS is an appropriate name for the former, and the latter entity should be identified as "cutaneous pseudolymphoma," or "mycosis fungoides-like lesions," as described in 1991.[20] Treatment The most important step in the management of AHS is to recognize the disorder and to discontinue the offending antiepileptic drug. Hospitalization is prudent for seizure prophylaxis and treatment. Interim anticonvulsant therapy, as previously discussed, should be guided by avoiding similar or interactive antiepileptic drugs [Table - 2]. Benzodiazepines may be used for short-term control. Severe cutaneous adverse reactions should be treated in consultation with a dermatologist or burn team depending on the extent of involvement. Continued enteral nutrition, intravenous fluid augmentation, pain relief, and ocular care in consultation with an ophthalmologist are essential.[21] Silver sulfadiazine and prophylactic antibiotics are not routinely recommended. The use of systemic corticosteroids has not been substantiated in randomized studies,[13],[16],[18] but has been considered the standard of care.[6] The preponderance of literature supports corticosteroid use in extensive cases, or cases with involvement of internal organs,[4],[18] although there are contrary data to suggest that only cutaneous manifestations are reversed.[16] When used, the most common dose is greater than 0.5 mg/kg per day,[13] or 60 mg of intravenous methylprednisolone every 6 hours.[22] Other immunomodulation (e.g. plasmapheresis, cyclophosphamide, cyclosporine, and intravenous immune globulin) has had anecdotal success.[23],[24],[25],[26] The prognosis of AHS varies widely in the literature. Mortality from AHS with associated hepatitis has been reported to range between 18% to 40%.[16] A recent review of hypersensitivity syndrome in general states that it is drug-induced in 90% percent of cases, with an overall mortality of 10%.[13] CONCLUSION The anticonvulsant hypersensitivity syndrome is an uncommon but serious disorder that should be recognized by physicians, who are very likely to be the first providers to encounter the initial manifestations of this disease. It is associated mainly with the aromatic anticonvulsants (phenobarbital, phenytoin, carbamazepine) but can be observed with the newer anticonvulsants. The diagnostic criteria are primarily fever, rash, and lymphadenopathy, but the gastrointestinal tract, lungs, and blood can be affected. Management emphasizes discontinuing the offending drug, supportive care in the inpatient setting because of the risk of seizure, and prudent consultation for specialty care. Corticosteroids should be considered in definitive drug eruptions only after infectious causes have been ruled out. REFERENCES
Copyright 2004 - Indian Journal of Dermatology, Venereology, Leprology The following images related to this document are available:Photo images[dv04016t1.jpg] [dv04016t2.jpg] |
| |||||||||

{kind=link}
{kind=link}