 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
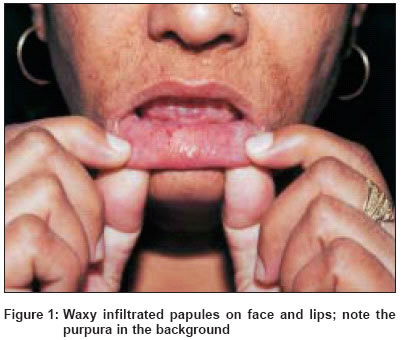
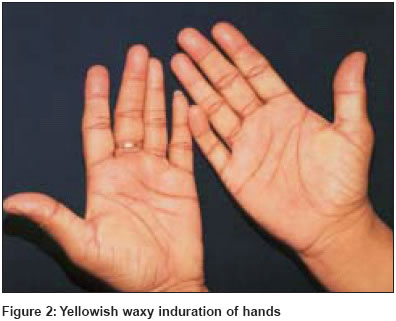
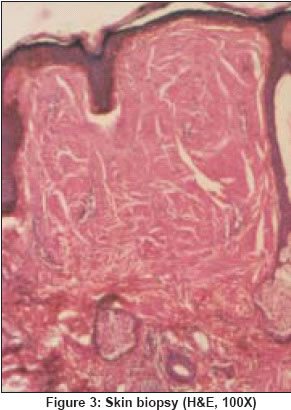
Indian Journal of Dermatology, Venereology, Leprology, Vol. 70, No. 4, July-August, 2004, pp. 263-265 Quiz Sclerodermoid hands with waxy papules on face Singhi MK, Gupta LalitK, Kachhawa Dilip, Bansal Mohit, Gupta Dhruv Department of Dermatology, Venereology and Leprosy, Dr. S. N. Medical College, Jodhpur (Rajasthan) Code Number: dv04093 A 46-year-old woman presented with asymptomatic, firm, translucent polygonal papules on the face [Figure - 1], mainly around the eyes and mouth, since 2 years. She also complained of symmetrical polyarthralgia involving the wrist, elbow, knee and ankle joints along with burning pain in the hands and feet. Both hands showed yellowish waxy papulonodular infiltrates [Figure - 2] and there was limitation of hand movements. The oral cavity showed papulonodular infiltrates with petechiae [Figure - 1]. The tongue was moderately enlarged. Systemic examination was normal other than mild hepatomegaly. Skin biopsy is shown in [Figure - 3]. What is the diagnosis? Answer: Primary systemic amyloidosis DISCUSSION The skin biopsy [Figure - 3] revealed fenestrated nodular deposits of eosinophilic, homogeneous material in the dermis staining metachromatically with crystal violet and revealed an apple green birefringence with alkaline Congo red staining. Bone marrow revealed plasmacytosis (10% plasma cells). Serum immunofixation electrophoresis [Figure - 4] revealed an IgG monoclonal band. Myeloma proteins were absent in the serum and urine. Amyloidosis is a metabolic disorder characterized by deposits of extracellular amorphous proteinaceous material in multiple organs impairing their functions. The disease is commonly classified as primary (occurring without antecedent or coexisting disease), secondary (associated with chronic inflammatory disease), myeloma associated, heredofamilial and localized.[1] Patients have a variable clinical picture depending on the predominant site of light chain deposition. Common clinical presentations include weakness, fatigue, peripheral neuropathy, periorbital purpura, dyspnea, pedal edema, syncope, light-headedness, hoarseness and dysphasia. A triad of carpal tunnel syndrome, macroglossia and cutaneous lesions is quite classical of plasma cell dyscrasia related systemic amyloidosis.[2] The frequent findings of abnormal plasma cells in the bone marrow, presence of mononuclear proteins in the serum or urine in most patients, and recognition that amyloid fibrils may be derived from immunoglobulin light chain fragments suggest that the cause of disease is almost certainly an underlying plasma cell dyscrasia.[2] About 40% of patients have monoclonal proteins on serum electrophoresis, 68% have a monoclonal spike on serum immunoelectrophoresis and 89% have a monoclonal spike on combined serum and urine electrophoresis.[3] The distinction between primary systemic amyloidosis and amyloidosis associated with multiple myeloma is difficult and may be of academic interest.[4] About 20% of patients with primary systemic amyloidosis meet the diagnostic criteria for multiple myeloma as determined by bone marrow biopsy, skeletal survey and elevated serum calcium level. One-fourth of the patients with amyloidosis in the absence of myeloma have greater than 10% of plasma cells in the bone marrow.[5] It is believed that the two diseases probably represent different poles of the spectrum of the same fundamental process. The prognosis of primary and myeloma associated amyloidosis is poor, the major cause of death being cardiac and renal failure. The average survival after diagnosis in most series is 1-3 years.[6] Melphalan, cyclophosphamide, prednisolone, colchicine and dimethylsulphoxide (DMSO) have been tried alone or in combination with variable success.[7] REFERENCES
Copyright 2004 - Indian Journal of Dermatology, Venereology, Leprology The following images related to this document are available:Photo images[dv04093f1.jpg] [dv04093f4.jpg] [dv04093f3.jpg] [dv04093f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}