 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
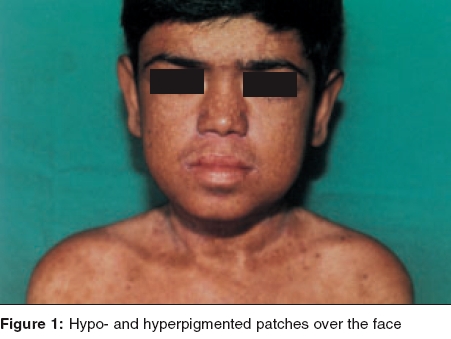
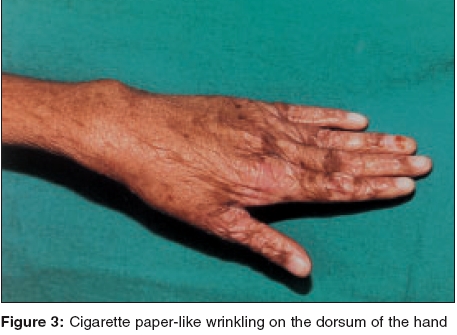
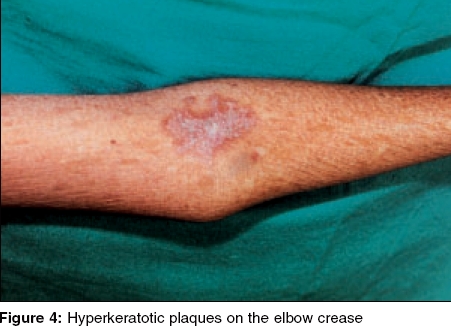
Indian Journal of Dermatology, Venereology and Leprology, Vol. 71, No. 5, September-October, 2005, pp. 348-350 Case Reports Kindler syndrome P. K. Kaviarasan, P. V. S. Prasad, Shradda, *P. Viswanathan Departments of Dermatology Venereology and Leprosy and *Pathology Rajah Muthiah Medical College and Hospital, AnnamalaiUniversity, India Correspondence Address:Plot no 88, AUTA Nagar, Sivapuri Post, Chidambaram-608002, Tamilnadu, prasaderm@hotmail.com Code Number: dv05114 ABSTRACT Kindler syndrome is a rare autosomal recessive disorder associated with skin fragility. It is characterized by blistering in infancy, photosensitivity and progressive poikiloderma. The syndrome involves the skin and mucous membrane with radiological changes. The genetic defect has been identified on the short arm of chromosome 20. This report describes an 18-year-old patient with classical features like blistering and photosensitivity in childhood and the subsequent development of poikiloderma. The differential diagnosis of Kindler syndrome includes diseases like Bloom syndrome, Cockayne syndrome, dyskeratosis congenita, epidermolysis bullosa, Rothmund-Thomson syndrome and xeroderma pigmentosum. Our patient had classical cutaneous features of Kindler syndrome with phimosis as a complication. Keywords: Kindler syndrome, Poikiloderma, Kindler-Weary syndrome INTRODUCTION Kindler syndrome is a rare hereditary disorder, which was first described in a 14-year-old girl by Kindler in 1954.[1] The patient had unusual blistering on the hands, arms, feet and legs and later developed photosensitivity and pigmentary changes. More than 120 cases have been reported since the original report by Kindler. Recently, 26 cases have been studied and reported,[2] which is the largest series of this syndrome. Clinical overlap with hereditary acrokeratotic poikiloderma and dystrophic epidermolysis bullosa may cause confusion.[3] Apart from the skin changes, changes in the oral and conjunctival mucosa, phimosis and radiological changes, namely a dome-shaped skull (turricephaly), rib and mandibular abnormalities have been reported.[4] The genetic defect of Kindler syndrome has been identified on the short arm of chromosome 20. The gene KIND I encodes a novel protein kindlin-I. This protein is involved in connecting the actin-cytoskeleton to the extracellular matrix. Histopathological examination shows features of poikiloderma like epidermal atrophy, hyper- and hypo-pigmentation and telangiectasia with hyperkeratosis, Areas of cleavage at or near the dermoepidermal junction may be present.[5],[6] We report a case of Kindler syndrome with phimosis as a complication.CASE REPORT An 18-year-old boy presented with a history of hypo-pigmented patches and photosensitivity since childhood. There was a history of acral blisters, which started on the fifth day of the neonatal period and persisted till 5 years of age. The patient had a history of recurrent respiratory infections and oral ulcers with halitosis. He also noticed generalized loss of sweating. He was born to consanguineous parents. His mother had vitiligo. On clinical examination, the patient weighed 25 kg and was short statured. His sexual maturity was in stage II instead of stage the IV, normal for his age. His IQ was normal. Cutaneous examination revealed multiple hypopigmented and a few hyperpigmented patches over the face [Figure - 1], neck, trunk and limbs. The trunk showed telangiectasia. The palms showed atrophic skin with loss of palmar creases [Figure - 2]. The dorsal aspect of the hands [Figure - 3] and feet showed atrophic scarring with shiny cigarette paper-like wrinkling. Hyperkeratotic plaques were seen on the flexures [Figure - 4]. The scalp hair were normal in color and growth pattern. Genital examination showed phimosis and scanty pubic hair. Oral mucosa revealed leukoplakia with few oral ulcers. Radiological screening revealed delayed epiphyseal fusion. Histopathological examination from a plaque revealed hyperkeratosis with flattened rete ridges and keratotic plugging. The dermis showed a lymphohistiocytic infiltrate in addition to melanophages. DISCUSSION Our patient presented with acral blisters in the neonatal period and childhood, diffuse poikiloderma, skin fragility and atrophic changes (cigarette paper-like wrinkled appearance of the skin), which were more prominent on the sun-exposed areas. Leukokeratosis of the oral mucosa and phimosis were additional features. The histopathology was consistent with poikilodermatous changes. The diagnosis of Kindler syndrome was made on the basis of these findings. Siegel et al have mapped the gene locus to band 20 p 12.3 by using linkage and homozygosity analysis in an isolated cohort of patients with Kindler syndrome.[7] Loss-of-function mutations were identified in the candidate gene KIND I. This gene encodes a protein, Kindlin I, which plays a regulatory role in inhibiting over-secretion of basement membrane components by basal keratinocytes at the dermo-epidermal junction. Kindler syndrome is suggested as the first skin fragility disorder caused by a defect in the actin-extracellular matrix (ECM) linkage rather than the keratin-ECM linkage. This syndrome has to be differentiated from Bloom syndrome, Cockayne syndrome, dyskeratosis congenita, epidermolysis bullosa, Rothmund-Thomson syndrome and xeroderma pigmentosum. Congenital bullous disorders may be differentiated by the spontaneous improvement of photosensitivity and blistering followed by development of poikiloderma and cutaneous atrophy seen in Kindler syndrome. Bloom syndrome shows telangiectasia, photosensitivity and erythema of the face and other sun exposed areas but not true poikiloderma. Short stature, recurrent infections and increased frequency of hematological malignancies are also present. Patients with Cockayne syndrome develop photo-distributed erythema, atrophy and hyperpigmentation. Other features are dwarfism, progressive pigmented retinopathy, deafness and bird-like facies, which are absent in Kindler syndrome. Rothmund-Thomson syndrome shows poikiloderma and photosensitivity in addition to sparse hair, hypogonadism and cataracts which are not seen in Kindler syndrome. Photosensitivity and the poikilodermatous changes found in xeroderma pigmentosum are found in patients with Kindler syndrome but acral bullae are not seen. In addition, patients with xeroderma pigmentosum experience early onset skin cancers and multiple neurological abnormalities. Dyskeratosis congenita presents with a triad of reticulated hyperpigmentation, nail dystrophy and leukoplakia. Pigmentary changes are not truly poikilodermatous and bullae are not prominent. Hereditary sclerosing poikiloderma of Weary is an autosomal dominant disorder characterized by progressive poikiloderma in flexural areas, sclerotic bands, poor dentition and calcinosis. Bullae and photosensitivity are not present. Treatment is mainly symptomatic with a focus on prevention of sundamage. Sun avoidance and photo-protection may delay the onset of poikiloderma. Complications range from secondary infections, urethral and esophageal stenosis, conjunctival scarring and periodontal disease. Patients may have a normal lifespan. Our patient presented with the classical features of Kindler syndrome. It was interesting to note that the patient had phimosis and there was a family history of vitiligo. Thappa et al reported Kindler syndrome in a 15-year-old Indian boy.[8] This case has been reported for its rarity. REFERENCES
Copyright 2005 - Indian Journal of Dermatology, Venereology and Leprology The following images related to this document are available:Photo images[dv05114f4.jpg] [dv05114f3.jpg] [dv05114f1.jpg] [dv05114f2.jpg] |
| |||||||||

{kind=link}
![Figure - 2]](/showimage?dv/photo/dv05114f2.jpg){kind=link}
{kind=link}
{kind=link}