 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
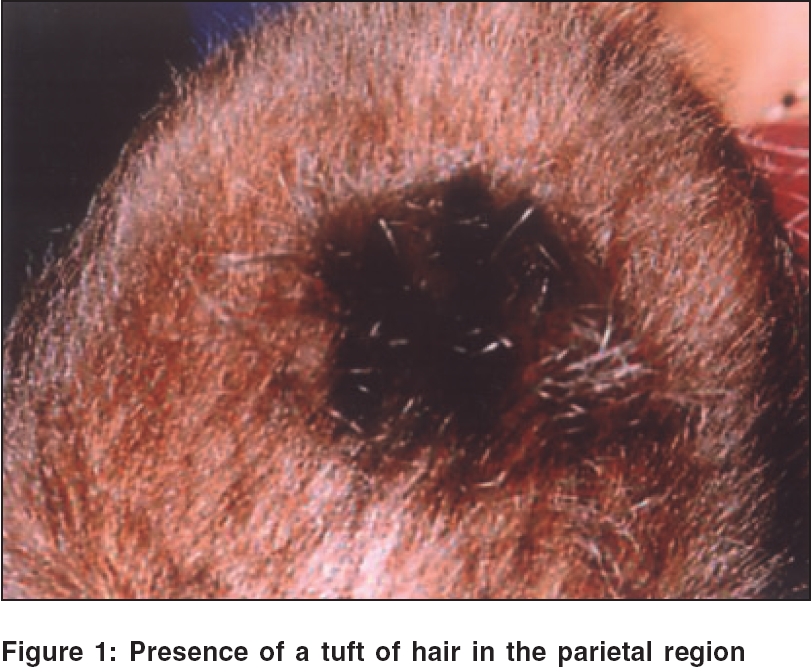
Indian Journal of Dermatology, Venereology and Leprology, Vol. 72, No. 2, March-April, 2006, pp. 147-149 Case Report Hurler syndrome with a tuft of hair Behera Binodini, Jena DK, Chhetia R, Vijayashree J Department of Skin, V. D. and Leprosy, SCB Medical College, Cuttack, Orissa Code Number: dv06045 Abstract A 2-year-old girl presented with coarse, thick hairy skin all over the body, a tuft of hair in the parietal region, coarse facial features and a prominent forehead with a large tongue, hepatosplenomegaly and skeletal deformities. Mucopolysaccharides excretion spot test of the urine was positive; and an assay for glycosaminoglycans in the urine was also high, which confirmed the clinical diagnosis of Hurler syndrome. We present this rare case to discuss the possibility of the association of mental retardation with a tuft of hair in this syndrome.Keywords: Bone marrow, Hurler syndrome, Mucopolysaccharidosis (MPS) - type I, Transplantation Introduction Hurler syndrome is an autosomal recessive disorder of mucopolysaccharide metabolism caused by a deficiency of the enzyme a-L-iduronidase.[1] The inability to degrade these macromolecules, which are ubiquitous, results in their storage in a variety of tissues including the liver, spleen, heart, connective tissue and others, resulting in premature death, usually by 10 years of age. These disorders display extensive genetic heterogeneity. In addition to somatic features, there may be severe mental retardation. Hurler syndrome represents the classical prototype of a mucopolysaccharide disorder, with a very low prevalence: 1:150,000 births. The diagnosis is usually made between the age of 6 and 24 months with evidence of coarse facial features, prominent forehead with large tongue, hepatosplenomegaly, corneal cloudiness, skeletal deformities, joint stiffness, short stature, acquired cardiomyopathy and progressive lenticular enlargement with increased intracranial pressure caused by communicating hydrocephalus. We present a case, which has not been reported so far, of mucopolysaccharidosis I (MPS-I) with a tuft of hair, in which the diagnosis was made on the basis of the clinical features and confirmed by enzymatic assays.Case Report A 2-year-old girl was brought to the Department of Skin and VD, S. C. B. Medical College, Cuttack, with complaints of coarse, thick hairy skin all over the body since birth. The antenatal, natal and neonatal histories were noncontributory. The child was born of a nonconsanguineous marriage and was the second child of the couple. There was no family history of a similar disorder or of neonatal deaths and miscarriages. According to the parents, all her milestones were delayed. On general examination, the girl was of low intelligence with a dull-looking appearance and was unable to speak and stand. She was 1 ft 20 cm tall and weighed 12 kg, with a large head and relatively short limbs. There was a tuft of hair in the parietal region [Figure - 1], widening of eyes with thick alae nasae, thickened lips, large protruding tongue and a short webbed neck. Her hands and feet were broadened and fingers were short, giving a crab-like appearance. The skin was thick and covered by coarse hair all over the body.[2],[3] She had a distended abdomen, umbilical hernia and hepatosplenomegaly. The respiratory and cardiovascular systems were normal. Neurological examination revealed mental retardation. Skeletal examination showed short stature with relatively short limbs. She had bilateral corneal clouding and hazy sclera. Hematological examination revealed normocytic, normochromic anemia. The biochemical profile was within normal limits. Routine urine examination was normal, but a spot test for mucopolysaccharides excretion in urine was positive. Urine assay for glycosaminoglycans showed a high level (5 mg/mmol). Discussion Hurler syndrome is an autosomal recessive disorder that has been described in families with a history of consanguinity.[1] Our patient was born of a nonconsanguineous marriage and without a family history of similar disease in the past two generations. She had a tuft of hair in the parietal region. Although rudimentary meningocele[4] and sequestrated meningocele[6] presenting with a scalp hair tuft have been reported, the presence of a tuft of hair has not been reported in Hurler syndrome to the best of our knowledge. To rule out any central nervous system disorder, any midline lesion with a hair tuft deserves proper evaluation including imaging studies.[4] A causal relationship has been proposed between anomalies of the central nervous system and cutaneous adnexal malformations.[6] In our view, although a midline tuft of scalp hair associated with central nervous system anomalies has not been reported as a feature of Hurler syndrome, its presence can be a useful clinical feature to initiate steps to rule out malformations affecting the central nervous system. Other clinical features that should arouse suspicion of Hurler syndrome include extensive Mongolian spots, inguinal hernia, broad costa and gibbus.[7],[8] Some other metabolic disorders that can manifest with abnormal hair characteristics like alopecia, hypopigmentation and abnormal texture[5] include phenylketonuria, congenital hypothyroidism, homocystinuria and biotinidase deficiency. For this reason, an assessment of the hair and scalp should be an integral part of every initial physical assessment of the newborn. Our patient had most of the features of Hurler syndrome except for its cardiovascular manifestations. The diagnosis was confirmed by a urine mucopolysaccharide excretion spot test and high-level glycosaminoglycans excretion in the urine. Hematopoietic stem cell transplantation is the treatment of choice for a child younger than 2 years of age suffering from Hurler syndrome with no or minimal central nervous system disease. Enzyme replacement therapy with a-L iduronidase is recommended for mild to moderate disease and for patients with neurological impairment.[9] To prevent mental retardation, a bone marrow transplant probably needs to be performed at a very early age. As bone marrow transplantation and enzyme replacement therapy were not possible in our setup, we advised regular follow-up and symptomatic treatment. Advances in stem cell transplantation and the use of recombinant enzymes necessitate early diagnosis in newborns with Hurler syndrome before the onset of irreversible tissue and organ damage. References
Copyright 2006 - Indian Journal of Dermatology, Venereology and Leprology The following images related to this document are available:Photo images[dv06045f1.jpg] |
| |||||||||

{kind=link}