 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
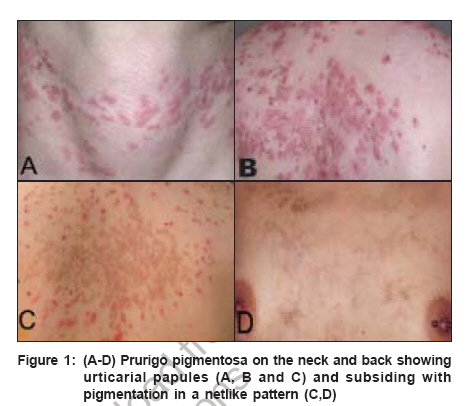
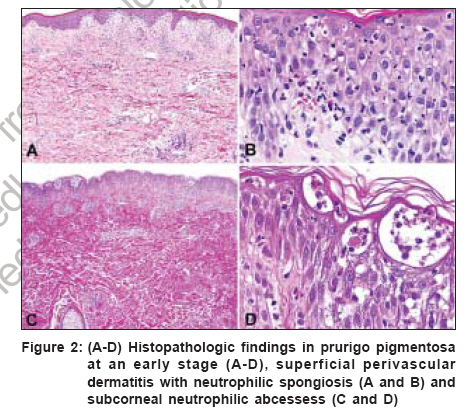
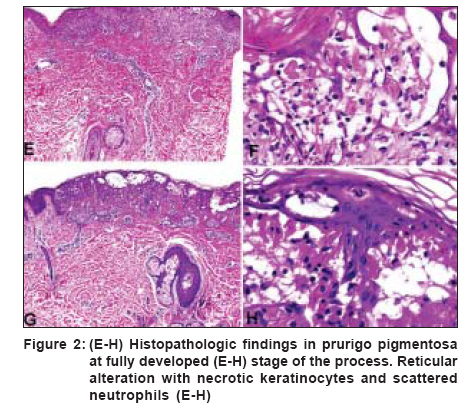
Indian Journal of Dermatology, Venereology and Leprology, Vol. 72, No. 6, November-December, 2006, pp. 405-409 View Point Prurigo pigmentosa: An underdiagnosed disease? Boer A, Asgari M Department of Dermatology and Dermatopathology, Dermatologikum, Hamburg Code Number: dv06144 Abstract Prurigo pigmentosa is a distinctive inflammatory disease first described by the Japanese dermatologist Masaji Nagashima in 1971. It is typified by recurrent, pruritic erythematous macules, papules and papulovesicles that resolve leaving behind netlike pigmentation. The disease is rarely diagnosed outside Japan, because clinicians outside Japan are not well conversant with the criteria for its diagnosis. Only one patient from India has been published previously under the diagnosis of prurigo pigmentosa, a hint that the disease may be under-recognized in India. We present an account of our observations in patients diagnosed with prurigo pigmentosa who were of five different nationalities, namely, Japanese, German, Indonesian, Turkish and Iranian. With this article we seek to increase awareness for the condition among dermatologists in India and we provide criteria for its diagnosis, both clinically and histopathologically.Keywords: India, Pigmentation, Pruritic exanthema, Prurigo pigmentosa, Reticulate hyperpigmentation Introduction Prurigo pigmentosa (PP) was first described by Nagashima et al . in 1971 and it is diagnosed most commonly in the Japanese population.[1],[2] PP is a rare inflammatory skin disease of unknown pathogenesis, typified by recurrent eruptions of pruritic erythematous macules and papules that resolve rather quickly, leaving behind netlike pigmentation. The disease is seen most commonly in young women from Japan and less than 40 non-Japanese patients have been reported.[1],[2],[3],[4],[5] It remains controversial whether PP really has a proclivity for the Japanese or whether it is often not diagnosed outside Japan because lack of awareness. Only one Indian patient with PP has been reported as of today. In 1992, Mittal and co-workers told of a 50-year-old diabetic female patient who presented with recurrent episodes of reticulate papular rash that resolved leaving a reticulate hyperpigmentation and that responded well to treatment with dapsone.[6] Recently, we were able to diagnose five patients of Iranian descent with PP, an ethnic population entirely unrelated to the Japanese and unknown previously to have a proclivity for PP.[7] That observation prompted us to conclude that PP might be more common outside Japan and is rarely diagnosed only because clinicians as well as histopathologists are not well aware of the criteria for diagnosis.[8] In our previous studies on the subject of PP, we elucidated the history of the disease and, based on our review of morphologic features in a large number of patients, we attempted to forge criteria for diagnosis of the disease both clinically and histopathologically.[3],[4],[5],[7],[8] Here, we present a summary of our observations. Clinical findings The eruption of PP is distributed symmetrically on the trunk, with predilection for the upper part of the back, the sacrum, abdomen and chest. Intermammary and submammary zones are involved more commonly than the clavicular site or the lateral side of the chest. When the back is affected, the shoulders and the upper part of the back are involved more often than the lower part or the lumbosacral region. Rarely, the forehead, the arms or the abdomen are caught up in the process. Mucous membranes are never affected. It is noteworthy that papules of PP tend to recur at the very same site where they have been present before. Individual lesions are patchy erythematous macules, urticarial papules and urticarial plaques at the beginning of an eruption, evolving to short-lived red papules and papulovesicles [Figure - 1]. Resolving lesions are crusted and scaly red papules and smooth surfaced pigmented macules. Lesions tend to be confluent and assume arcuate and reticular shapes. Often, lesions of different ages are seen together and the pattern of lesions is netlike. At the outset, lesions are intensely pruritic and signs of excoriation are common. Histopathology In an early lesion of PP the pattern of the infiltrate is that of a sparse perivascular and interstitial dermatitis that most commonly is rather superficial but that may, occasionally, be mid-dermal or even deep, though it always is top heavy [Figure - 2a-d, Figure - 2e-h]. The spectrum of changes histopathologically in evolving lesions of PP ranges from scattered neutrophils in the epidermis to poorly formed microabscesses and to large collections of neutrophils beneath the cornified layer. The degree of spongiosis ranges from slight to marked. As a rule, spongiosis is more prominent in evolving lesions than in resolving lesions. Occasional necrotic keratinocytes are present in conjunction with scattered neutrophils and spongiosis in the epidermis. Lymphocytes are sprinkled along the dermo-epidermal junction at the same time when neutrophils dominated the upper layers of the epidermis. In a fully developed lesion, the infiltrate assumes a patchy lichenoid pattern and lymphocytes in the dermis are more numerous [Figure - 2a-d, Figure - 2e-h]. In the epidermis ballooning is more prominent than spongiosis and lymphocytes in the lower part of the epidermis are increased in number. Necrotic keratocytes disposed as solitary units or in small clusters are a common finding, but necrosis en masse is present sometimes, thereby causing a blister to come into being. In the upper part of an epidermis with changes such as these, neutrophils and their nuclear dust may be recognizable within aggregations of necrotic keratocytes and also beneath the cornified layer in the viable epidermis adjacent to necrotic epidermis. The cornified layer still is orthokeratotic and basket-woven at that stage of the process. In a resolving lesion, a sparse infiltrate of lymphocytes is present in the upper part of the reticular dermis and the papillary dermis. Necrotic keratinocytes may be found in the basal layer. The cornified layer may show parakeratosis and scale crust. Melanophages in the papillary dermis and the upper part of the reticular dermis range from few to many. Often lesions show signs of rubbing and scratching in those specimens features typical of the disease may hardly be recognized with surety. Differential diagnoses Diseases to be considered in the differential diagnosis clinically at an early stage of the process are early manifestations of dermatitis herpetiformis, linear IgA dermatosis and acute lupus erythematosus, even though the trunk-centered distribution of lesions and the reticular arrangement of individual lesions in PP allow differentiation. At a resolving stage of the process, PP may be confused with reticular pigmented papules of confluent and reticulated papillomatosis of Gougerot and Carteaud, but in contrast to those lesions, pigmented macules of PP are not keratotic. Diseases that have to be considered in the histopathologic differential diagnosis vary according to the stage of the disease process. Early in the course, urticaria, evolving leukocytoclastic vasculitis, dermatitis herpetiformis, linear IgA dermatosis, acute lupus erythematosus, eruptive psoriasis or dermatophytosis may be considered because of the predominance of neutrophils in the infiltrate, but epidermal changes typical of PP, such as spongiosis joined by ballooning, scattered neutrophils in the epidermis and necrotic keratinocytes in the absence of features diagnostic of any of the conditions mentioned usually enable differentiation. A fully developed lesion of PP has to be differentiated from erythema multiforme and Mucha Haberman disease. Collections of neutrophils beneath the cornified layer as well as a few eosinophils in the infiltrate are clues to the diagnosis of PP. At a late stage of the process, histopathologic features of PP are indistinguishable from any other disease that resolves with postinflammatory hyperpigmentation. In clinicopathologic correlation, however, the disease may still be diagnosed with certainty because of the typical netlike distribution of pigmentation centered on the trunk. Clinical course, Treatment and Etiology Lesions of prurigo pigmentosa resolve in a matter of days as crusted and scaly papules, which are slightly reddish in color. Crusted and scaly lesions are present for a few days only and eventually subside leaving behind pigmented macules, which may persist for months. That is why crusted and scaly papules as well as pigmented macules are seen much more commonly in patients with prurigo pigmentosa than are urticarial papules and papulovesicles. Many patients with PP experience spontaneous resolution of eruptions after a few weeks, recurrences occurring only months or years later. Other patients, when left untreated, experience recurrences every month over years. Several medications are available for treatment of PP, minocycline and dapsone being used most commonly. Both medications have in common antibiotic as well as antiinflammatory effects and they are especially effective in the inhibition of migration of neutrophils, a mechanism which may explain their efficacy in PP.[3],[4],[5] A recent report told of a favorable response of PP to isotretinoin, the mechanism of action of this medication being opaque.[9] Various mechanisms have been suggested to cause PP, such as friction, contact allergy, sensitivity to sunlight, endocrinologic alterations such as diabetes mellitus and metabolic disorders such as ketosis, but none of them proved to be identifiable consistently. No association with other diseases or with significant results of ancillary laboratory procedures such as levels of autoantibodies or studies by immunofluorescence has been identified yet. Cause and pathogenesis of PP remain to be elucidated. Comment Criteria for the diagnosis of PP are summarized in [Table - 1]. Certain clues to the diagnosis of PP are listed in [Table - 2]. For clinicians unfamiliar with PP it is necessary to be fully aware of the dynamics of an eruption lasting no more than a week from beginning to end. By way of clinicopathological correlation, the disease can be diagnosed with confidence at any stage of the process, but histopathological features are diagnosable with specificity much more easily at the beginning and at the summit of the eruption, than at a time when the lesions resolve. If a patient is agreeable, at least two biopsy specimens should be obtained, one of which should be taken from an urticarial lesion that has been present less than two days. Urticarial lesions are usually present for less than 48h and, therefore, a dermatologist should take the biopsies on the day when a patient is seeking consultation for the first time rather than giving him or her a new appointment for taking the biopsy a few days later. If a specimen shows changes in the cornified layer such as parakeratosis or scale crust in a patient with clinical signs of prurigo pigmentosa, a resolving lesion has been biopsied and changes diagnostic of the disease cannot be expected in such a specimen. Lesions that show scratch marks should not be biopsied because excoriated lesions cannot be diagnosed with confidence. The report by Mittal and co-workers in 1992 demonstrates that PP exists in India. The fact that no second patient from India has been reported during the last 14 years can be interpreted in various ways. Either PP is extraordinarily rare in India or it is rarely diagnosed because colleagues are unaware of the condition or not confident with the criteria for its diagnosis. We hypothesize that PP might be more common in India and, with this article we hope to increase awareness of PP among colleagues in India. References
Copyright 2006 - Indian Journal of Dermatology, Venereology and Leprology The following images related to this document are available:Photo images[dv06144f2a-d.jpg] [dv06144t1.jpg] [dv06144f1.jpg] [dv06144f2e-h.jpg] [dv06144t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}