 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
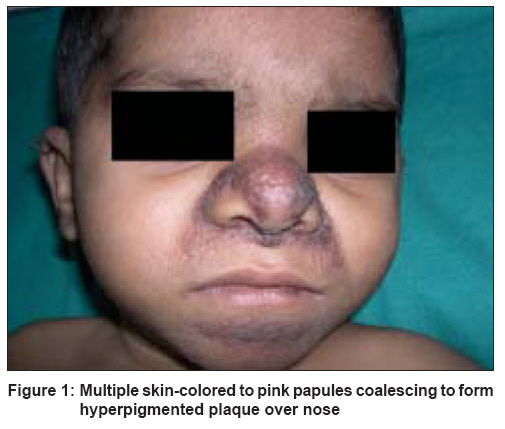
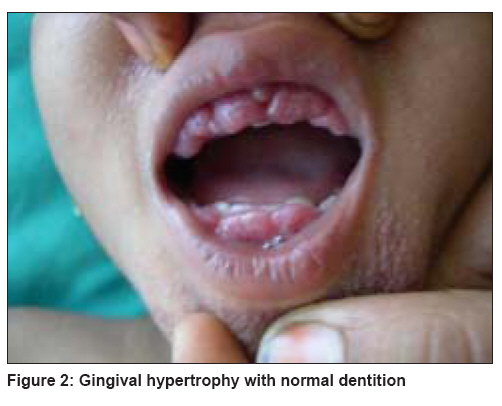
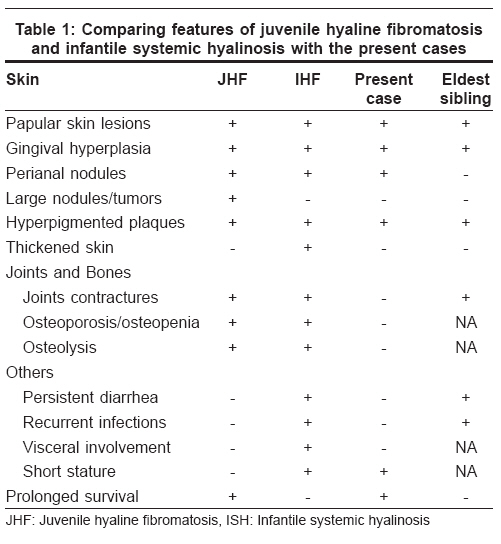
Indian Journal of Dermatology, Venereology and Leprology, Vol. 74, No. 4, July-August, 2008, pp. 371-374 Case Report Juvenile hyaline fibromatosis and infantile systemic hyalinosis: Divergent expressions of the same genetic defect? Dhingra Mandeep, Amladi Sangeeta, Savant Shankar, Nayak Chitra Department of Derrmatology, T.N. Medical College and B.Y.L. Nair Ch. Hospital, Mumbai Central, Mumbai - 400 008 Code Number: dv08160 Abstract We describe here a three year-old girl with classic clinical and histological features of juvenile hyaline fibromatosis. We found a history of similar skin findings in her eldest sister, in whom the disorder took a rapidly progressive and fatal course in the second year of life, suggesting either a very severe form of juvenile hyaline fibromatosis, or the possibility of infantile systemic hyalinosis. The similarities and differences between these two described types of hyalinoses have been reviewed in reference to the present report.Keywords: Allelic disorders, Fibromatosis, Hyalinosis Introduction Hyalinoses are rare autosomal recessive disorders in which there is an accumulation of amorphous hyaline material in the skin and other organs. Two distinct forms have been described: juvenile hyaline fibromatosis (JHF) and infantile systemic hyalinosis (ISH). Juvenile hyaline fibromatosis is characterized by papulonodular skin lesions, gingival hyperplasia, joint contractures, and osteolytic bone lesions. Skin lesions may consist of multiple large tumors, commonly found on the scalp and the neck, and small pearly, pink papules and plaques on the trunk, chin, ears, and around the nostrils. [1] The clinical features of infantile systemic fibromatosis include a diffuse thickening of the skin, papulonodular skin lesions, hyperpigmentation over the metacarpophalangeal joints of the hands and malleoli, osteoporosis of bones, bone fractures, short stature, persistent diarrhea, and failure to thrive. [1] Case Report A three year-old girl was brought by her mother with complaints of asymptomatic, red colored, raised lesions prevalent since she was one year of age, on her nose, perioral area and ears. She had delayed milestones with restriction of speech to a few meaningful words only. She was unable to stand without support and had recurrent episodes of diarrhea. She had no history of joint contractures, convulsions or hoarseness of voice. She was born at full term by normal delivery following an uneventful pregnancy. Her parents had third degree consanguinity and were both unaffected. The middle sister aged five years, was unaffected by the disorder. Her eldest sister however, born six years earlier, had developed identical, papular skin lesions on the face early in the neonatal period which gradually increased in size and number. As per the mother′s history, she had shown restriction of movement of the right upper limb since early infancy. She did not have thickened skin but was malnourished and had been chronically unwell with multiple hospital admissions for recurrent episodes of pneumonia and diarrhea. She had died at age of 1½ years of some cardiac problem, the details of which were unavailable. Examination of our index case showed that she was short for her age (height 75 cm). Her weight was 10 kg implying grade-I malnutrition. The child was responsive to her surroundings, but could speak only a few meaningful words and was unable to support herself in the standing position due to muscle weakness. Cutaneous examination revealed the presence of 1-5 mm, multiple, skin-colored and pink papules over the face and ear lobes. They were darker, clustered together and confluent on the mid-face, especially the nose, nasolabial folds and chin [Figure - 1]. Tiny papules were visible on the eyelids. Ulceration and discharge was evident from the nasal lesions, suggesting secondary infection. The trunk and limbs were spared. There was a single, large, soft, reddish nodule, 5 cm in size in the perianal area. Examination of the oral cavity revealed gingival hyperplasia; dentition was normal. Systemic examination did not reveal any abnormality. The patient was found to be moderately mentally retarded with an IQ of 50. A complete hemogram, renal function tests, liver function tests, blood sugar and electrolytes were within normal limits. X-rays of the chest and bones were normal. Electrocardiograpy and 2D-echocardiography did not reveal any cardiac abnormality. Skin biopsy of the lesions showed a normal epidermis with deposits of amorphous, eosinophilic hyaline material throughout the upper and mid dermis. There was an abundance of stroma with a relative paucity of cellular elements. Numerous spindle-shaped, fibroblast-like cells were seen in the vicinity of the deposits [Figure - 2]. On special staining, the deposits were seen to be Periodic Acid-Schiff (PAS)-positive, Congo red-negative and Alcian blue-negative. The child was diagnosed as a case of juvenile hyaline fibromatosis. Although we had not examined the eldest sister, her features as per her mother′s history are suggestive of a severe and rapidly fatal form of JHF or possibly ISH [Table - 1]. The parents were counselled about the progressive nature of the disease and the 25% chance of development of disease in future offspring. The patient is currently undergoing physiotherapy for the improvement of muscle strength. Discussion Juvenile hyaline fibromatosis (synonyms: juvenile hyalinosis, fibromatosis hyalinica multiplex, Murray-Puretic-Drescher syndrome) [2] is a rare, autosomal recessive, hereditary disease with distinct clinical and histopathological features. A survey of literature revealed < 70 cases reported worldwide [3] and only a handful of case reports from India. [3],[4] There is no sex predilection. The baby is normal at birth but abnormalities develop during the first two years and are relentlessly progressive. [2] Gingival hypertrophy often develops in the first year of life. The papules are distributed about the nose, behind the ears, in the genital area and on the thighs. Joint contractures often develop. Several large nodules and tumors develop between two and five years of age. Infection often results from ulceration of the lesions. [2] Adolescent patients are usually disfigured and bedridden due to their contractures. [5],[6] Partial disease expression is common. [7] Infantile systemic hyalinosis (infantile hyaline fibromatosis, infantile hyalinoses) [2] is a disorder clinically similar to juvenile hyaline fibromatosis, but with far more severe joint involvement, joint contractures and thickened skin. Infants are affected within the first few weeks or months of life. Recurrent purulent infections, diarrhea and severe osteoporosis are observed in the first year of life. Severe joint limitation and pain lead to immobility and respiratory insufficiency. Feeding problems, malnutrition and protein-losing enteropathy are caused by the thickening and hyaline infiltration of intestinal walls. Death occurs secondary to sepsis with renal, respiratory and heart failure, usually by the age of two years. [8],[9] It appears that both the conditions are linked to a disorder of the synthesis of glycosaminoglycans with resulting abnormality in collagen synthesis. The gene locus has been found on chromosome 4q21. [2] Mutations in the capillary morphogenesis gene-2 ( CMG2 ) have been found recently. [10] Histology shows the deposition of an amorphous, eosinophilic hyaline material in the extracellular spaces of the dermis around the blood vessels. There is an abundance of stroma with a relative paucity of cellular elements. The dermis contains a few inflammatory cells and widely scattered spindle cells disposed in a densely eosinophilic background. [2] The deposits are PAS-positive and Alcian blue-negative. Differential diagnosis of hyaline deposits in the skin includes disorders such as amyloidosis, lipoid proteinosis, and Waldenstr φm`s macroglobulinemia. They can also be seen in gouty tophi (if fixed in an aqueous fixative), colloid milium and at corticosteroid injection sites. Perivascular hyaline deposits are a feature of porphyria, especially erythropoietic protoporphyria. [11] Both the disorders are allelic [10] and intermediate phenotypes between these two disorders have been described. It has been suggested that both disorders constitute parts of the same disease. [1] Patients with severe forms of JHF with persistent diarrhea and death in early infancy due to infections, have been reported. [1] Postmortem findings in another case with typical lesions of JHF strongly resembled those of ISH. In our reported family, the eldest sibling had features suggesting a diagnosis of a severe and rapidly fatal form of JHF or possibly ISH. This occurrence of two different ends of the spectrum of disease in siblings reiterates the view that both these conditions constitute parts of the same disease. Other differential diagnoses include neurofibromatosis, gingival hyperplasia, nodular amyloidosis, congenital generalised fibromatosis, Winchester syndrome, and lipoid proteinosis. [3] Inborn metabolic disorders such as Farber`s disease, mucopolysaccharidoses and I-cell disease should also be considered in differential diagnosis. Gingival fibromatosis is not associated with cutaneous lesions typical of hyalinosis. Winchester syndrome is always associated with corneal clouding. Lipoid proteinosis can be differentiated on clinical grounds as well as by the demonstration of deposition of a amorphous, eosinophilic, hyaline material within the dermis with an onion skin arrangement around blood vessels. Inborn errors of metabolism can be differentiated on the basis of histological and biochemical findings. [2] No specific treatment is available. Early surgical excision is recommended by some authors in JHF to prevent the appearance of new lesions, [12] although excision may be followed by recurrences. [5] Intralesional steroids may reduce the size of early lesions. Excision is indicated only for those lesions that either present a significant cosmetic problem or produce some degree of functional impairment. Capsulotomy of joints may show some temporary, beneficial effect; radiotherapy is ineffective. [12] Gingival overgrowth may be treated with partial gingivectomy. Oral D-penicillamine has been used in some cases. [1] Therapeutic trials with dimethyl sulfoxide, ketotifen, and calcitriol have been given in individual cases. [12] Although the prognosis is better in JHF, the patients are left with deformities and joint contractures. References
Copyright 2008 - Indian Journal of Dermatology, Venereology and Leprology The following images related to this document are available:Photo images[dv08160t1.jpg] [dv08160f1.jpg] [dv08160f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}