 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
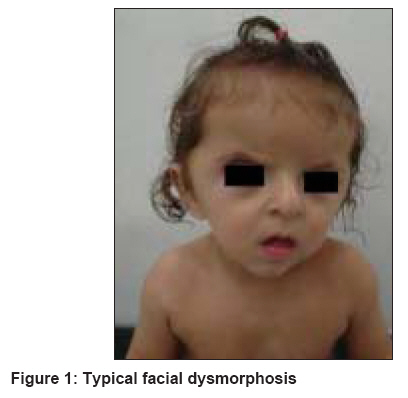
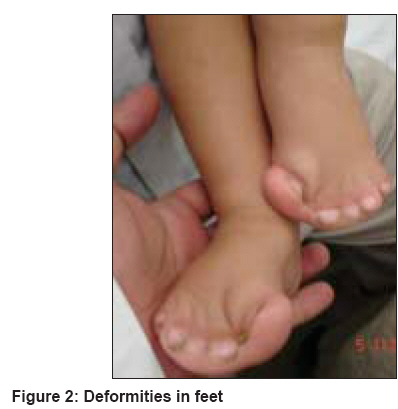
Indian Journal of Dermatology, Venereology and Leprology, Vol. 74, No. 4, July-August, 2008, pp. 395-396 Letter To Editor Brachycephaly and syndactyly: Apert's syndrome De Dipankar, Narang Tarun, Kanwar AmrinderJ, Dogra Sunil Department of Dermatology, Postgraduate Institute of Medical Education and Research, Chandigarh-160 012 Code Number: dv08172 Sir, Apert′s syndrome (acrocephalosyndactyly type 1) is a rare malformation syndrome first described by Wheaton in 1894, and later by Apert in 1906. [1] It is characterized by craniosynostosis associated with maxillary hypoplasia, symmetric syndactyly of the hands and feet, and other systemic malformations including mental retardation. Acne is the most common cutaneous association of Apert′s syndrome. An eight-month-old boy born of nonconsanguineous parents presented with multiple congenital anomalies. The mother′s antenatal period was uneventful, with no history of any infectious illness, medication(s) exposure or addiction during that period. Family history was non-contributory. The patient′s anomalies included craniofacial abnormality and syndactyly of all four limbs. The child had a typical facial appearance with a short, wide head (brachycephaly), frontal bossing, retruded (sunken) mid-face, depressed nasal bridge, upward slanting of the eyes, low-set ears and a low posterior hairline [Figure - 1]. Examination of the upper and lower limbs showed symmetrical syndactyly leading to the fusion of all the fingers and toes [Figure - 2]. The child had both growth and mental retardation. Apert′s syndrome is specifically related to the paternal age effect having a frequency of one in every 65,000 live births; the affected patients generally do not survive beyond infancy. [1] In those patients who reach adolescence, acne is an important cutaneous manifestation. Most cases are sporadic, although autosomal dominant inheritance as well as germinal mosaicism have been reported. [2] The syndrome is caused by nucleotide alterations resulting in amino acid substitutions, leading to a mutation in the fibroblast growth factor receptor-2 (FGFR-2) gene mapped to chromosome 10q26. [3] Premature fusion of the bones is responsible for bony abnormalities; premature fusion of coronal sutures lead to brachycephaly. Other craniofacial abnormalities include a prominent forehead with skin wrinkling, a broad cranium, and a flat occiput. The shortened bony orbit leads to hypertelorism, proptosis and strabismus. Additional features include a short broad nose with a bulbous tip, micrognathia, and a cleft palate. Intracranial anomalies include megalocephaly, progressive hydrocephalus, hypoplastic white matter, and agenesis of the corpus callosum and limbic structures, leading to cognitive impairment. Cardiac abnormalities including atrial and ventricular septal defects and renal anomalies such as hydronephrosis occur in about 10% of these patients. The cutaneous abnormalities reported are acne, hyperhidrosis and oculocutaneous albinism. Acne is now known to represent the dermatological hallmark of Apert′s syndrome; it usually appears between the age of nine and 12 years. The acneiform lesions involve not only the face and upper trunk, but also affect the forearms, buttocks, and thighs. Oily skin is striking by the time of adolescence but the etiology of these acneiform lesions remains controversial. Increases in circulatory androgens or the density of sebaceous gland androgen receptors have not been documented. End-organ androgen metabolism defects, structural malformation of pilosebaceous apparatus and a role of FGFR-2 in regulating androgen sensitivity of the pilosebaceous unit have been suggested hypotheses for the acneiform lesions. [4] The skin, eyes and hair may show pigmentary dilution. Other occasional cutaneous abnormalities include interrupted eyebrows, forehead wrinkling, paronychial infections, skin dimpling over the knuckles, shoulders and elbows, as well as lateral plantar hyperkeratosis. There is no definitive treatment for this syndrome. The prognosis varies from child to child and a multidisciplinary approach is recommended. References
Copyright 2008 - Indian Journal of Dermatology, Venereology and Leprology The following images related to this document are available:Photo images[dv08172f2.jpg] [dv08172f1.jpg] |
| |||||||||

{kind=link}
{kind=link}