 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
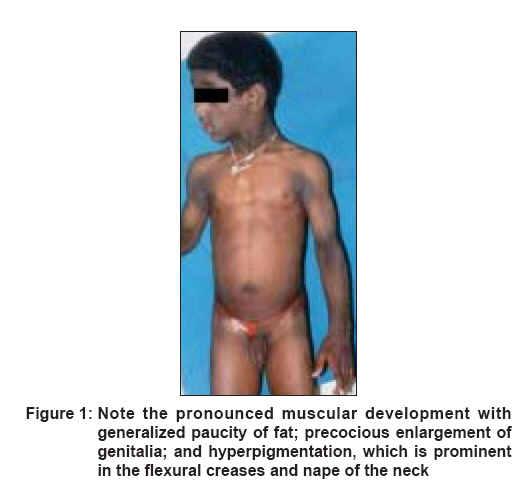
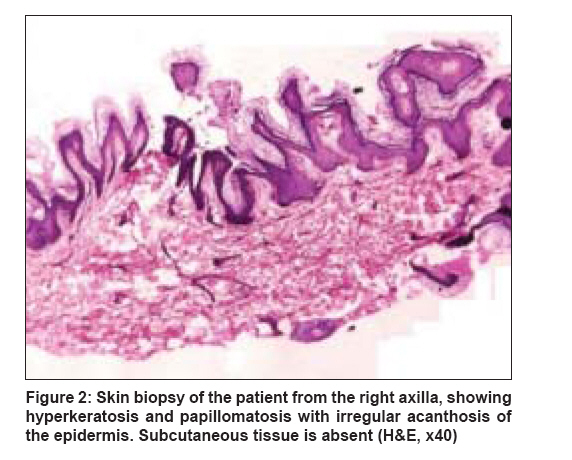
Indian Journal of Dermatology, Venereology and Leprology, Vol. 74, No. 6, November-December, 2008, pp. 644-646 Case Report Berardinelli-Seip syndrome in a 6-year-old boy Babu Priya, Sharma Rakesh, Jayaseelan Elizabeth, Appachu Divya Department of Dermatology, St. John's Medical College Hospital, Bangalore Code Number: dv08260 Abstract A 6-year-old boy presented with abnormal habitus since birth, delayed language development, history of frequent falls since 9 months, and fever since 1 week. He was found to have hyperandrogenic features, generalized paucity of fat, generalized muscular overdevelopment, and brownish pigmentation over the flexural creases. Skin biopsy demonstrated features suggestive of acanthosis nigricans with an absence of subcutaneous tissue. After further investigation, a diagnosis of Berardinelli-Seip syndrome with bilateral pneumonia and generalized tonic clonic seizures was made. Clinical features, histopathology, differential diagnosis, and prognosis of this rare disorder have been discussed.Keywords: Acanthosis nigricans, Berardinelli-Seip syndrome, Congenital generalized lipodystrophy, Lawrence-Seip syndrome Introduction Berardinelli-Seip syndrome, or ′Berardinelli-Seip congenital lipodystrophy′ (BSCL), is a rare autosomal recessive disorder characterized by lipoatrophy, hypertriglyceridemia, hepatomegaly, and acromegaloid features. Other features include mental retardation; hypertrichosis; precocious puberty; hypertrophic cardiomyopathy; and insulin-resistant diabetes mellitus, which usually manifests in the second decade of life. The prevalence of BSCL is estimated to be 1 per 0.2 million in Lebanon, 1 per 0.5 million in Portugal, 1 per 1 million in Norway, and 1 per 12 million in USA. [1] Approximately 120 patients of various ethnic backgrounds have been reported. [2]Case Report A 6-year-old boy, the younger of two siblings from a third-degree consanguineous marriage, presented with abnormal facies and body habitus since birth. Parents were concerned that he looked much older than children of his age and had not developed language skills. On his first presentation at our hospital, it was recorded that he had a history of frequent abnormal falls since 9 months, an episode of generalized tonic clonic seizures (GTCS) lasting for less than a minute 2 weeks back, and fever since 1 week. He was a normal full-term baby delivered by lower segment cesarean section (LSCS), with no prenatal, perinatal, or postnatal complications and with up-to-date immunizations. On general examination , the child was found to have dysmorphic features like acromegaloid facies, generalized lipoatrophy, hypertrichosis, luxuriant hair growth over back and forehead, precocious enlargement of genitalia with a penile length of 6 cm (expected size according to Tanner stage 1 - penis of 3 cm or less), umbilical hernia with slightly protuberant abdomen, pronounced muscular development, and generalized hyperpigmentation which was more prominent in the flexural creases and nape of the neck [Figure - 1]. His weight was 24 kg (118% of expected), and height was 127 cm (116% of expected). His temperature, respiratory rate, pulse rate, and blood pressure recordings were within normal limits. Cervical lymph nodes were palpable and oral thrush was present. Systemic examination revealed liver palpable 3 cm below the right costal margin, which was firm; spleen was not palpable and there was no free fluid. An ejection systolic murmur in the left parasternal area and apex was diagnosed. Respiratory system was normal. He was found to have subnormal intelligence with no signs suggestive of meningeal irritation. Fundus examination was normal. Laboratory investigations revealed hemoglobin of 13.7 gm%, total WBC - 175,000; N 67 L 14 M 4 Meta 8 Myelo 7 ; smear for malarial parasites - negative, blood culture - no growth; Widal titers not suggestive of enteric fever; Mantoux test was negative; urinalysis showed occasional WBCs, urine sugar measured by Benedict′s test was 1+. The lipid profile was as follows: serum high density lipoproteins (HDL) - 31 g/dL, serum low density lipoproteins (LDL) - 111 mg/dL, serum cholesterol -175 mg/dL, and serum triglycerides -166 mg/dL. Total serum proteins were 8.3 mg/dL, with albumin of 4.9 mg/dL, and alkaline phosphatase (ALP) was 380 U/L. The fasting blood sugar was 147 mg/dL, and the glucose tolerance curve was abnormal. Serum insulin level was 12.3 microunit/mL (normal: 5-35 microunit/mL). Luteinizing hormone (LH) and follicle stimulating hormone (FSH) values were (0.6/0.6) and (4.8/2.15) respectively, in 30 minutes/60 minutes after GnRH injection. Chest x-ray revealed bilateral patchy pneumonia, and skeletal survey of the pelvis and upper femur showed advanced bone age. X-ray of the skull (occipital mental view) showed features suggestive of fibrous dysplasia. A skin biopsy taken from the right axilla revealed features of acanthosis nigricans. Subcutaneous tissue was absent [Figure - 2]. KOH of the oral thrush was positive for Candida sp. The child was treated with antibiotics for the pneumonia and sodium valproate for the seizure disorder. The elevated sugar level was managed with diet restrictions and oral pioglitazone, 15 mg once daily. With this therapy, the pneumonia resolved, his seizure disorder was controlled and his hyperglycemia was better controlled.Discussion This disease is called ′Berardinelli-Seip syndrome′ after Berardinelli from Brazil, who described the first patients, [3] and it was confirmed by Seip from Norway in 1959. It is characterized by the almost total lack of subcutaneous adipose tissue, severe diabetes mellitus, no ketosis/ketonuria and insulin resistance; the latter usually develops in the second decade of life. [3],[4] Other characteristics include gum hypertrophy, hepatomegaly affecting liver functions, hypertriglyceridemia, acanthosis nigricans, arterial hypertension, hirsutism, nephropathy, and cardiac hypertrophy. [5],[6] The case described above had many of these features. Generalized lipodystrophy is often classified into a congenital and an acquired form, with Lawrence-Seip syndrome being acquired generalized lipodystrophy and Berardinelli-Seip syndrome (BSS) being congenital generalized lipodystrophy. BSS is transmitted as an autosomal recessive trait and is linked to two genetic loci, on chromosomes 9q34 (AGPAT2 gene) and 11q13 (BSCL2 gene).[7] Mutations in the AGPAT2 and BSCL2 genes are known to be associated with BSS type 1 and type 2 respectively. Type 1 seems to be less severe; with some cases of type 2 resulting in premature death, which can occur as early as the first year of life. Approximately 80% of individuals with mutations in BSCL2 have mild-to-moderate intellectual impairment, whereas only 10% of individuals with mutations in AGPAT2 have intellectual impairment. Mutations in AGPAT2 may cause congenital lipodystrophy by inhibiting/reducing triacylglycerol synthesis and storage in adipocytes. It is also likely that reduced AGPAT2 activity could increase tissue levels of lysophosphatidic acid, which may negatively affect adipocyte functions. [8] It has been shown that lipodystrophy is induced by a compound similar to cachectin [9],[10] (tumor necrosis factor). This has powerful inhibitory effects on lipoprotein lipase and causes fat depletion and hyperlipidemia when injected in animals. The pathogenesis of acanthosis nigricans when associated with Insulin resistance is explained by epithelial proliferation mediated through insulin, which stimulates keratinocytes and fibroblasts, both of which express insulin and insulin-like growth factor receptors. [11] The skin changes like hypermelanosis, hypertrichosis, and acanthosis nigricans found in our patient were similar to that found by Reddy et al. [12] in 1986. These children can have associated immune deficiency, as described by Kher et al. [10] in 1990. This may have been the cause of oral candidiasis and lower respiratory infection in our patient. Restriction of fat to between 20% and 30% will help to control the triglyceride levels, and the patient should be followed up from time to time in the diabetology clinic. [1] A new option for therapy is leptin, an adipocyte hormone, which may improve insulin resistance, hyperglycemia, dyslipidemia, and hepatic steatosis. [13] Leptin is the first of a group of adipocyte-secreted hormones to be used clinically. [14] Its long-term efficacy in 15 patients with generalized lipodystrophy was evaluated. [15] Leptin has been judged the first novel, effective, long-term treatment for severe forms of lipodystrophy. Patients with the congenital form of Lawrence-Seip syndrome tend to survive into young adulthood or early middle age. A common cause of death has been GI hemorrhage caused by esophageal varices in association with hepatic failure. Renal complications are also a frequent cause of death. Acknowledgement We wish to express our thanks to Dr Betty Alexander, Assistant Professor, Department of Pathology, St. John′s Medical College, Bangalore and Dr. Chitra Dinaker, Assistant Professor, Department of Paediatrics, St. John′s Medical College Hospital, Bangalore for their assistance.References
Copyright 2008 - Indian Journal of Dermatology, Venereology and Leprology The following images related to this document are available:Photo images[dv08260f1.jpg] [dv08260f2.jpg] |
| |||||||||


{kind=link}
{kind=link}