 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
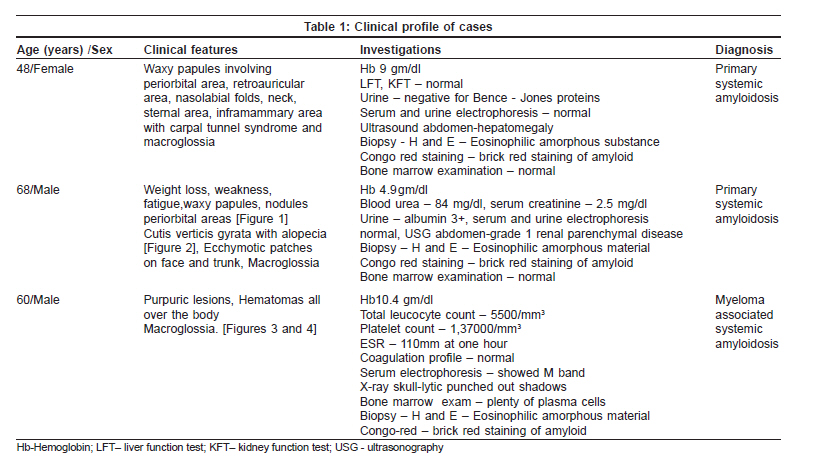
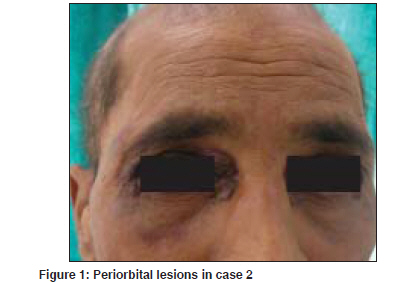
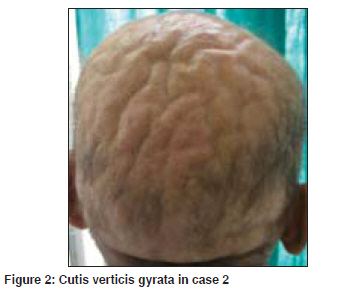
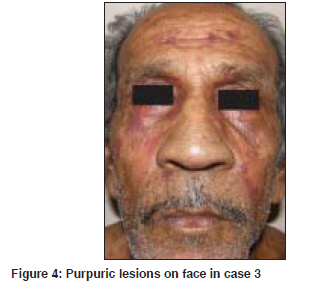
Indian Journal of Dermatology, Venereology and Leprology, Vol. 75, No. 4, July-August, 2009, pp. 394-397 Case Report Primary systemic amyloidosis: Three different presentations Saoji Vikrant, Chaudhari Sanjiv, Gohokar Dilip Department of Dermatology, Jawaharlal Nehru Medical College, Sawangi, Meghe, Wardha Code Number: dv09151 PMID: 19584467 Abstract Primary systemic amyloidosis is a rare disease. We report three cases of primary systemic amyloidosis, one case with multiple myeloma and two cases without any hematological abnormality. Purpuric lesions were the only presenting symptoms of the patient with multiple myeloma and only on investigation, myeloma was detected. Bone marrow biopsy and serum and urine electrophoresis were normal in remaining two cases. These two patients presented with typical waxy lesions on face. Cutis verticis gyrata was present in one case and carpal tunnel syndrome was seen in other case as an additional diagnostic clue. Macroglossia was present in all three cases. Diagnosis was confirmed in all three cases by biopsy using haematoxylin and eosin staining and Congo red staining. Polarized microscopy was not done because of unavailability.Keywords: Primary systemic amyloidosis, Myeloma, Idiopathic Introduction Amyloidosis is a disease caused by the deposition of insoluble abnormal fibrillary protein known as amyloid in the extracellular space. [1] Amyloid deposition may be localized to one organ system or it may involve multiple organs depending on which amyloidosis is classified as localized or systemic. Localized cutaneous amyloidosis, which includes macular amyloidosis and lichen amyloidosis, is a benign common disease without systemic involvement. Skin may also be involved in systemic amyloidosis. Systemic amyloidosis is classified into primary, secondary and familial. [2] Primary systemic amyloidosis may be idiopathic or myeloma-associated. Secondary systemic amyloidosis is associated with many chronic inflammatory disorders. Skin involvement is common in primary amyloidosis whereas secondary amyloidosis rarely involve skin. [3] Systemic amyloidosis is usually a disease of the elderly. All forms of systemic amyloidosis are a rare disorder. We report three cases of primary systemic amyloidosis, one with myeloma-associated primary amyloidosis and two without any plasma cell abnormality. Case Reports Clinical findings in these three cases are described in the [Table - 1]. All three cases presented with asymptomatic skin lesions. The first two cases first presented to other departments and then was referred to dermatology OPD for skin lesions. In all three cases, because of typical presentations, diagnosis of primary systemic amyloidosis, was suspected clinically and confirmed by histopathology. Polarized microscopy was not done. Macroglossia was present in all three cases and was very important clinical feature to suggest the possibility of systemic amyloidosis. Classical features like carpal tunnel syndrome, macroglossia and skin lesions were present in first case only. Hepatomegaly was seen in case 1. Evidence of renal failure like raised blood urea and serum creatinine along with urine albumin was present in case 2. Out of these three cases, hematological abnormality (multiple myeloma) could be detected only in case 3. In cases 1 and 2, bone marrow examination, serum and urine electrophoresis were normal. In case no. 3, diagnosis of multiple myeloma was made due to bone marrow examination and serum electrophoresis. X-ray skull also showed lytic shadows suggestive of multiple myeloma. Discussion Amyloidosis is a disease caused by deposition of highly insoluble fibrous protein amyloid in the tissues. A protein is called amyloid if it gets mis-folded to form a beta- pleated sheet structure. Polypeptide chain gets folded to form a secondary structure of the protein molecule. Helical structure and pleated structure are two important secondary structures of protein molecule. The amyloid protein has a beta-pleated sheet structure, which makes it highly insoluble and resistant to proteolytic digestion and hence difficult to remove from the tissues. [3] About 25 different proteins are known to produce amyloid fibrils in human, most of them are constituents of plasma. [4] These normally soluble precursor proteins, due to some unknown reason, get mis-folded and forms a beta-pleated sheet structure and becomes amyloid. [4] Inherited amyloidosis is due to mutation in certain precursor protein, which makes them susceptible to mis- folding. In case of primary systemic amyloidosis, the amyloid is derived from monoclonal immunoglobulin light chain and is called as AL amyloid where L stands for light chain of immunoglobulin molecule. In case of secondary amyloidosis which is associated with many chronic inflammatory diseases, amyloid fibrils are derived from cleavage fragment of the circulating acute phase reactant serum amyloid A protein (SAA), hence called as AA amyloid. Serum amyloid A protein is synthesized in liver during inflammation. [2] In localized cutaneous amyloidosis, amyloid is derived from keratin released from apoptotic keratinocytes. [1] The reason that many diverse conditions are associated with amyloidosis may be because each of these conditions results in excessive production of proteins that are prone to mis-folding. [2] In multiple myeloma-associated AL amyloidosis, precursor light chains of immunoglobulin (Bence Jones protein) are produced in large quantity by malignant plasma cell clone and can be detected in serum or urine by electrophoresis. Multiple myeloma is a malignancy of plasma cell. Amyloidosis develops in about 15% of patients of myelomatosis. [3] Majority of patients of AL amyloidosis do not have obvious B-cell/ plasma cell neoplasm (idiopathic). These patients might have underlying B-cell dyscrasia in which production of abnormal protein, rather than production of tumor masses, is the predominant manifestation. [2] In one study from Lebanon, of 39 cases of systemic amyloidosis, 21 were of AL type and out of these 21 cases of AL amyloidosis, 9 (43%) were associated with multiple myeloma and 12 (57%) were idiopathic. [5] In an autopsy study from Germany of 43 cases of systemic amyloidosis, AL amyloidosis was found in 10 (23%) of which 50% were associated with myeloma and another 50% idiopathic. [6] Although different types of amyloid are associated with distinct clinical picture, all amyloid share a certain common features: [7]
Deposition of amyloid in tissues leads to distortion of tissue architecture, organ enlargement (organomegaly) and organ dysfunction. Amyloid deposition can occur in any organ. Primary systemic amyloidosis (AL) is known for highly varied clinical manifestation [8] as evident from our three cases. Cutaneous involvement is seen in 40% patients with AL amyloidosis. [3] Cutaneous manifestation depends upon the site of amyloid deposited. Amyloid deposition in superficial dermis produces shiny waxy translucent papules and common sites for predilection are eyelids, retro-auricular areas, neck, axilla, as present in our cases 1 and 2. Amyloid deposited around pilosebaceous unit leads to the destruction of hair, producing alopecia. Diffuse infiltration of scalp skin results in the enlargement of skin which gets thrown into longitudinal folds resembling cutis vertices gyrata. Alopecia with cutis verticis gyrata was present in our case 2. Diffuse infiltration of large area of skin may simulate scleroderma. Infiltration of nail matrix by amyloid may produce ridging, splitting and brittleness of nail plate. [3] Amyloid infiltration of vessel wall causes capillary wall fragility, which leads to purpura and ecchymosis after a minor trauma or even spontaneously. Periorbital area is one of the common sites of expression of purpura. The capillary fragility may be demonstrated by pinching the skin. Ecchymotic lesions were present in cases 2 and 3, and the only presenting symptoms in our case 3. Purpuric lesions with normal platelet count and normal coagulation profile should suggest the possibility of capillary fragility. Senile purpura also is due to capillary fragility but is commonly seen on extremities and very rarely on face. Amyloid deposition in tongue leads to macroglossia. Tongue is diffusely enlarged and firm and there may be tooth indentation along its lateral border. Amyloidosis is the commonest cause of macroglossia in adults. [3] Macroglossia if severe might lead to dysphagia. Macroglossia with tooth indentation was present in all our three cases. Hepatomegaly occurs in 50% of patients and splenomegaly in 10%. Hepatomegaly was present in our case 1. Cardiac involvement leads to conduction defects, arrhythmias, congestive cardiac failure and may account for 40% of deaths. None of our three patients had cardiac involvement as indicated by normal ECG and X-ray chest. Carpal tunnel syndrome is seen in up to 25% of patients of primary systemic amyloidosis [3] as was present in our case 1. Amyloid infiltration might occur in peripheral nerves leading to thickening of nerves and resulting neuropathy which may mimic Hansen′s disease. Renal involvement presents with proteinuria and renal failure. It is one of the bad prognostic indicator and was present in our case 2 as indicated by proteinuria and USG finding. Of our three patients, case 3 had multiple myeloma and in other two patients, we could not find any hematological abnormality as indicated by normal bone marrow biopsy and negative serum electrophoresis. All three patients were diagnosed as having primary amyloidosis on clinical ground. In all these three cases, diagnosis was confirmed by demonstration of amyloid in skin biopsy. Clinically, it is difficult to distinguish primary, secondary or familial form of amyloidosis. Immunohistochemical staining using commercially available antisera is useful for classifying the type of amyloid deposited in tissues. [9] Biopsy is very important for the diagnosis. Hematoxylin and eosin staining suggests the possibility of amyloidosis but Congo red staining confirms the diagnosis. Congo red staining results in a brick red color of amyloid when seen under ordinary light and under polarized light shows classical green birefringence. Unfortunately, polarized microscopy is not easily available in developing country like India. In systemic amyloidosis, amyloid deposits are seen in dermis, subcutaneous tissue and blood vessels, where as in localized cutaneous amyloidosis, deposits are seen only in papillary dermis; subcutaneous tissues and blood vessels are not involved. Prognosis in AL amyloidosis is poor and major causes of death are cardiac and renal failure. The median survival of patients with myeloma-associated amyloidosis is five months and 2.1 years for patients with primary systemic amyloidosis. [3] Prognosis depends upon the extent of involvement. Treatment of amyloidosis is aimed at reducing the supply of precursor proteins. [1] In AL amyloidosis, the precursor is immunoglobulin light chain produced by B lymphocytes/plasma cells hence treatment with cytotoxic agents like melphalan and prednisolone that reduces plasma cell proliferation is useful. [1] Chemotherapy will be useful only when precursors are supplied by plasma cells like AL amyloidosis. In localized cutaneous amyloidosis, such as lichen amyloidosis and macular amyloidosis, where precursors are derived from keratinocytes and not from plasma cells, alkylating agents or any other chemotherapeutic agents will not be beneficial and may be harmful. [10] These cases of systemic amyloidosis are presented for its rare occurrence. High index of suspicion is necessary for the diagnosis of such rare cases. [Figure - 1], [Figure - 2], [Figure - 3], [Figure - 4] References
Copyright 2009 - Indian Journal of Dermatology, Venereology and Leprology The following images related to this document are available:Photo images[dv09125t1.jpg] [dv09125f3.jpg] [dv09125f2.jpg] [dv09125f4.jpg] [dv09125f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}