 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
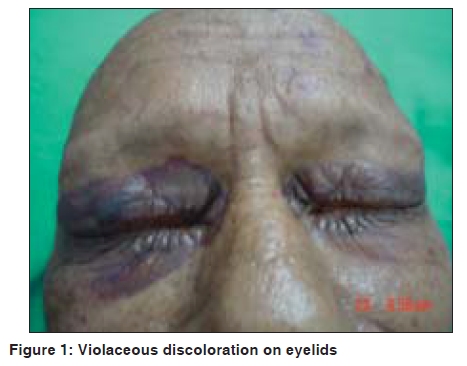
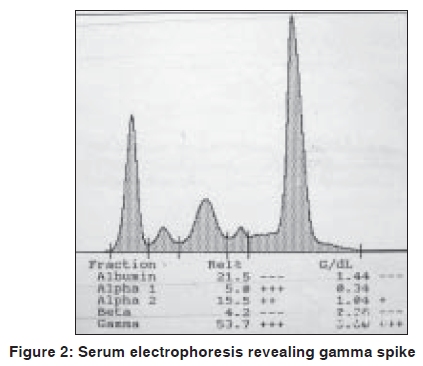
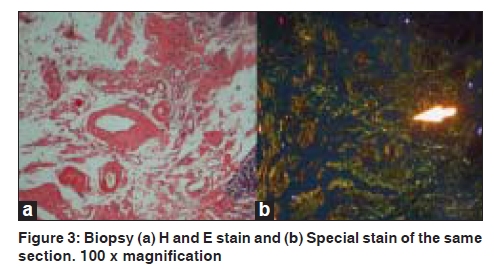
Indian Journal of Dermatology, Venereology and Leprology, Vol. 75, No. 5, September-October, 2009, pp. 549-550 Quiz Violaceous discoloration around the eyes Biju Vasudevan, M. P. S. Sawhney Department of Dermatology, Base Hospital, Delhi Cantonment, New Delhi-110 010, India Code Number: dv09187 PMID: 19736459 DOI: 10.4103/0378-6323.55425 A 57-year-old male presented with dark pigmentation around the eyelids, of one-year duration. He also had joint pain and weight loss for the same duration. A general physical examination revealed pedal edema and pallor, while a systemic examination showed ascites, pleural effusion (right), and asterixis. A dermatological examination revealed violaceous pigmentation on both eyelids [Figure - 1]. The patient also had similar lesions on the neck, axilla, and upper chest. Macroglossia was present as was alopecia. Investigations revealed turbidity in the urine with proteinuria of 3+ and high 24-hour urinary protein levels. Blood urea and serum creatinine were raised. An X-ray of the skull showed multiple translucencies and that of the hips and spine showed osteosclerosis. Serum electrophoresis revealed M protein with gamma spike [Figure - 2]. Histopathological findings were as shown in [Figure - 3]. What is your Diagnosis ? Hematoxylin and eosin stained section of the biopsy specimen revealed a homogenous eosinophilic, hyaline material deposited around blood vessels while a Congo red stain of the same section revealed classic apple green birefringence confirming the diagnosis of amyloidosis. Discussion Amyloidosis is a rare systemic disorder of protein metabolism with progressive extracellular deposition of an insoluble fibrillary protein, disorganization of tissue architecture, and subsequent organ dysfunction.[1] It can be systemic or strictly cutaneous. Systemic amyloidosis may be primary, myeloma-associated, or secondary. Amyloidosis is presently biochemically classified based on fibril properties, such as, light chain or transthyretin associated. In primary and myeloma-associated amyloidosis, the fibril protein is deposited as an immunoglobulin light chain or light chain fragment: so they are known as light chain amyloidosis (AL). AL amyloidosis is rare, with an incidence of less than one per 100,000, but mucocutaneous lesions may occur in up to 40% of these patients. The broad spectrum of skin manifestations in systemic amyloidosis may vary from purpuric lesions to sclerodermal changes. Purpura, petechiae, and ecchymoses occur due to intracutaneous hemorrhage resulting from amyloid infiltrating and weakening the blood vessel walls and are particularly common in skin folds such as the eyelids, axillae, umbilicus, and the anogenital area. Periorbital purpura may arise after coughing, sneezing, performing proctoscopy or the Valsalva maneuver. The periorbital lesion has to be differentiated from the heliotrope rash of dermatomyositis. Gottron's sign and Gottron's papules help the differentiate this condition from the cutaneous features of amyloidosis. Other conditions that can rarely produce similar eyelid lesions are systemic lupus erythematosus and drug reactions to hydroxyurea, niflumic acid, tryptophan, d-penicillamine, and practolol. Multiple myeloma is a monoclonal B-cell neoplasm characterized by the autonomous proliferation of immunoglobulin secreting plasma cells that are capable of synthesizing amyloidogenic light chains, resulting in AL amyloidosis. It has been estimated that 15% of the patients suffering from multiple myeloma demonstrate amyloidosis. [2] The amyloid deposits can be identified on the basis of their apple-green birefringence under a polarized light microscope after staining with Congo red, and the presence of rigid, non-branching fibrils, 7.5 to 10 nm in diameter, on electron microscopy. [3] The recent development of a fully quantitative serum Fibril light chain assay, to sensitively monitor the AL fibril precursor protein has been a huge advance in the diagnosis and management of systemic AL amyloidosis. Treatments with colchicine, high-dose steroids, and alpha-interferon have shown promising results in slowing the progression of amyloidosis. Preliminary research has shown that high dose melphalan, iododoxorubicin followed by bone marrow transplantation, or autologous stem-cell transplantation may be future treatments of choice in amyloidosis associated with multiple myeloma.[4] In recent times, anti-amyloidogenic or anti-protein misfolding therapies that target the process of protein misfolding and aggregation have been under development and appear promising. The prognosis of combined amyloidosis and multiple myeloma is improving with the new therapeutic options. [5] The greatest prognostic factor is the stage of the disease at the time of initial treatment, demonstrating that early diagnosis is of paramount importance. In this case we had a patient presenting with cutaneous signs of systemic amyloidosis, who was found to have multiple myeloma on investigation, paving way for his early treatment, thus emphasising the importance of cutaneous signs in diagnosing systemic disorders. References
Copyright 2009 - Indian Journal of Dermatology, Venereology and Leprology The following images related to this document are available:Photo images[dv09187f1.jpg] [dv09187f2.jpg] [dv09187f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}