 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
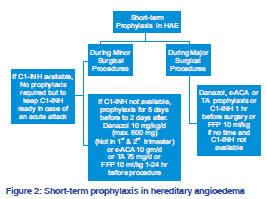
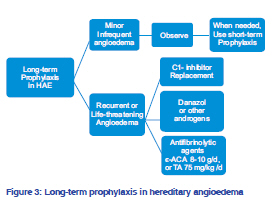
Indian Journal of Dermatology, Venereology, and Leprology, Vol. 77, No. 5, September-October, 2011, pp. 621-624 Focus Hereditary angioedema: An update Pramod Kumar Nigam Department of Dermatology and STD, Pt. JNM Medical College, Raipur - 492 001, Chhattisgarh, India Correspondence Address: Code Number: dv11184 DOI:10.4103/0378-6323.84093 Hereditary angioedema (HAE) is a rare, autosomal dominant disorder of C1 inhibitor (C1-INH) deficiency manifested with any combination of cutaneous angioedema (painless, nonpruritic, nonpitting swelling of the submucosal, dermal, or subcutaneous tissue), severe abdominal pain, or acute airway obstruction. HAE occurs in 1 in 50,000 individuals.[1] It is a potentially life-threatening disorder, with some studies reporting lifetime disease-associated mortality rates up to 33%, in hospitalized patients.[1] Any race can be affected, with no reported bias in different ethnic groups. CLINICAL AND MOLECULAR CLASSIFICATION OF HEREDITARY ANGIOEDEMA Three types of HAE are recognized, designated as types I-III. The classic forms, types I and II, result from mutations in the gene encoding C1 inhibitor protein (C1- INH, SERPING1),[2] whereas type III involves mutations in the F12 gene, encoding coagulation factor XII (Hageman factor).[3] Type I HAE is characterized by low plasma levels of a normal C1-INH protein, type II HAE is characterized by the presence of normal or elevated levels of a dysfunctional C1-INH, whereas type III HAE has been recently identified as an estrogen-dependent form of angioedema occurring mainly in women with normal functional and quantitative levels of C1-INH. Type I HAE is the most common form and accounts for about 85% of cases. Type III HAE is relatively rare and is expressed mainly in females as it is often associated with increased circulating estrogen. PRECIPITATING FACTORS AND DISEASE ASSOCIATIONS Precipitating factors for types I and II HAE may include trauma, anxiety, menstruation, infection, exercise, alcohol consumption, and stress. Medications such as estrogen, ACE inhibitors, and angiotensin II receptor antagonists have also been shown to induce attacks.[4] Most attacks, however, do not have a trigger. Precipitating factors for type III HAE include high estrogen conditions such as pregnancy and individuals on oral contraceptives or hormone replacement therapy.[3,5] Systemic lupus erythematosus (SLE) and other autoimmune diseases, such as glomerulonephritis, rheumatoid arthritis, thyroiditis, Sjögren syndrome, and pernicious anemia, may be associated with HAE.[4] Additionally, individuals with Helicobacter pylori infection have been found to be more symptomatic than those who are not infected. MOLECULAR PATHOLOGY The gene for C1-INH (SERPING1) has been mapped to chromosome 11 (q12-q13.1). More than 180 different mutations have been described in types I and II HAE. Typically missense mutations lead to decreased C1-INH levels and type I HAE, whereas deletions, frameshift or splice site mutations result in decreased activity of C1-INH and underlie type II HAE.[2] The F12 gene maps to chromosome 5 in HAE III. In type III HAE, the C1-INH protein is both qualitatively and functionally normal. More recently, mutations in factor XII have been identified in some, but not all patients. These factor XII mutations allow for the inappropriate activation of the kinin cascade. Pathophysiology The mechanism of angioedema in the absence of C1- INH appears to be secondary to increased levels of bradykinin.[8] C1-INH is a regulatory enzyme (serpin) that modulates the complement, coagulation, kinin, and fibrinolytic pathways. The angioedema resulting from both C1-INH and F12 mutations involves the same bradykinin-mediated pathway. The deficiency of C1-INH results in excessive formation of kallikrein resulting in higher turnover of kinins. During contact activation, F12 is activated, which leads to increased levels of kallikrein production through the cleavage of plasma prekallikrein. Kallikrein then catalyzes bradykinin synthesis from high molecular weight kininogen.[2] Bradykinin is a very potent vasodilator and plays a pivotal role in the development of angioedema.[9] Bradykinin acts through the bradykinin receptor 2 (B2) present on vascular endothelial cell membrane resulting in vasodilatation and increased vascular permeability. Clinical features The clinical features of acute attacks of type I-III HAE are indistinguishable. Typically, the onset of HAE occurs in childhood, although the disease often becomes more severe in adolescence. Symptoms relate to acute edema in three main sites: subcutaneous tissues, viscera, and the upper airway.[2] Clinically, an acute episode of HAE is associated with swelling that worsens over 12-24 hours and then resolves within 72 hours but may last 3-5 days.[10] Acute abdominal pain, nausea, and vomiting are the dominant symptoms in 25% of patients with HAE, but they rarely occur with other forms of angioedema. Rare symptoms include urinary retention due to mucosal edema of the bladder or urethra. Prodromal symptoms such as severe headache, visual disturbances, sudden mood changes, anxiety, or exhaustion often precede the onset of acute angioedema. Diagnosis and differential diagnosis HAE is currently recognized as a genetic disorder with autosomal dominant transmission. The presence of a family history of angioedema assists in diagnosis. Complement studies, including C4 and C1-esterase inhibitor, both antigenic and functional C1-INH are the most commonly used tests in the diagnosis of HAE. Analysis of C1q can help differentiate between HAE and acquired angioedema (AAE) caused by C1-INH deficiency. Genetic tests would be particularly helpful in patients with no family history of angioedema, which occurs in about half of the affected patients and in patients whose C1q level is borderline and does not differentiate between HAE and AAE. Specific genetic tests used in HAE include fluorescence-assisted mismatch analysis, specific PCR amplification and real-time PCR analysis, chemical cleavage of mismatches, denaturing high-performance liquid chromatography and gradient gel electrophoresis, and single-stranded conformational analysis. Measuring autoantibodies against C1-INH would also be helpful, but the test is available in research laboratories only. Simple complement determinations are appropriate for screening and diagnosis of the disorder. HAE must be distinguished from acquired angioedema (AAE) with C1 esterase inhibitor deficiency, angiotensin-converting enzyme inhibitorinduced angioedema, and the much more common histaminergic angioedema, occurring with or without urticaria. The accurate diagnosis of type I or II HAE can be made by laboratory assessment of C1-INH levels or activity (in type III HAE C1-INH levels are normal). Management of acute attack of angioedema in adults with hereditary angioedema angioedema, intensive support may be necessary, including intravenous fluids [Figure 1]. In cases of serious laryngeal edema causing respiratory = INH concentrates[11] (20 U/kg) or kallikrein inhibitor or, if unavailable, fresh-frozen plasma (FFP). Subcutaneous administration of the bradykinin receptor antagonist icatibant, a selective bradykinin B2 receptor antagonist, peptidomimetic drug, is another therapeutic possibility. New bradykinin receptor antagonists such as anatibant, MEN16152, and FK5657 are under development.[12] Another new therapeutic option in treating acute episodes of HAE is ecallantide, a kallikrein inhibitor.[13] A recombinant C1- INH RhucinTM is at an advance stage of development. Mild abdominal symptoms can be treated with antispasmodics but abdominal obstruction requires a C1-INH or bradykinin inhibitor.[14] Systemic corticosteroids, epinephrine, and antihistamines are not beneficial. In HAE type III, infusion of C1-INH has proven to be ineffective.[15] but bradykinin receptor antagonist might prove effective.[12] Prophylaxis against hereditary angioedema As short-term prophylaxis before surgical procedures, 500-1000 U of C1-INH concentrate 60 min before the procedure or 600 mg of oral danazol for 2 days can be administered 2-5 days before the procedure.[16] For long-term prophylaxis, administration of anabolic androgens, such as 200 mg of oral danazol, or alternatively stanozolol or oxandrolone, is recommended. An alternative drug, more effective in HAE type III, is tranexamic acid, an antifibrinolytic, that competitively inhibits the activation of plasminogen to plasmin [Figures 2 and 3]. Management of hereditary angioedema in women Hormonal factors such as puberty, contraception, and pregnancy are known to play a significant role in the precipitation or worsening of the HAE in women.[17] For short-term prophylaxis in females, three options, C1-INH concentrate, tranexamic acid, and attenuated androgens, are available.[17] Antifibrinolytic (tranexamic acid) are often the first best choice for HAE women because of good tolerance. Attack rates can increase during pregnancy and C1-INH concentrate and tranexamic acid can be used in pregnant women.[17] Attenuated androgens for long-term prophylaxis are found to be effective but dosedependent side effects appear more often in female patients and should not be used in the case of pregnancy and lactation.[17] The changes of attack frequency during the last trimester of pregnancy are of potential prognostic value. Pregnancy with a fetus affected by HAE-C1-INH was associated with a significant (P = 0.039) increase in the number of edematous attacks experienced by the mother during the third trimester.[18] The incidence of breast cancer is not higher than that in the rest of the population. Tamoxifen should not be used as it may worsen symptoms.[19] Plasma derived C1 inhibitor (pdC1-INH) is the safest prophylactic agent during pregnancy.[20] Danazol should be avoided in the first two trimesters of pregnancy.[20,21] Management of hereditary angioedema in children concentrate or 10 ml/kg FFP can be given.[22,23] For less severe abdominal attacks, tranexamic acid (12-25 mg/ kg, to a maximum of 1.5 g) is an alternative option.[22] For short-term prophylaxis before surgical procedures, 25 U/kg of C1-INH concentrate or 10 mg/kg/day danazol can be given. Alternatively, tranexamic acid 12-25 mg/kg may be administered 5 days before and after surgery.[22,24] For long-term prophylaxis, tranexamic acid 12-25 mg/kg/day orally in divided doses as first line treatment or alternatively, oral danazol in a dose up to 200 mg/day or 400 mg weekly may be used. Androgen therapy is not recommended for children but has been used in the prepubertal setting.[20,21] pdC1-INH may be the safest long-term approach in children also.[20,21] CONCLUSION Hereditary angioedema is a rare but important disease for clinicians and dermatologists to recognize and manage. The diagnosis of the type of angioedema is important not only for identification of therapeutic options but also for family planning and counseling. Accurate diagnosis can be made by estimating complement levels. To date, genetic testing for HAE is available only in the research laboratory because of the multitude of different mutations leading to the disorder. About half of the patients do not have a family history of disorder. If the family history is positive, the diagnosis is easier to make. Management of HAE involves treatment of acute attacks as well as prophylaxis. Newer treatments target the bradykinin B2 receptors or inhibit kallikrein activity as specific therapies for HAE. REFERENCES
Copyright 2011 - Indian Journal of Dermatology, Venereology, and Leprology The following images related to this document are available:Photo images[dv11184f3.jpg] [dv11184f2.jpg] [dv11184f1.jpg] |
| |||||||||

![[Figure 1]](/showimage?dv/photo/dv11184f1.jpg){kind=link}
{kind=link}
{kind=link}